臨床試験
海外第Ⅲ相試験3)
3ヵ月以上安定したIgG補充療法を受けているPID患者を対象とした先行治療からの切り替え試験
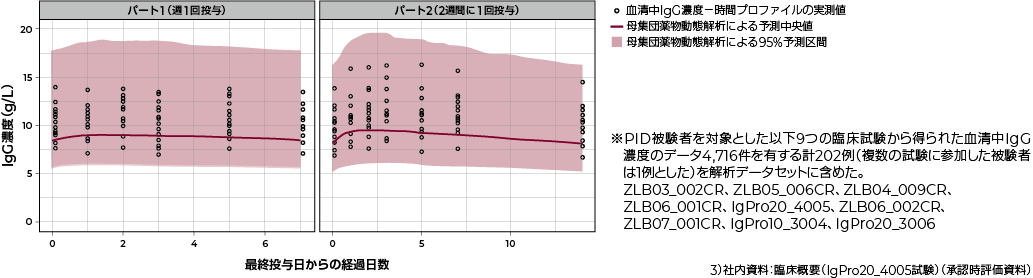
3)社内資料:臨床概要(IgPro20_4005試験)(承認時評価資料)
試験概要
目的
[主要目的]
ハイゼントラの2週間に1回投与の忍容性及び安全性を評価する。
[副次目的]
- ハイゼントラの2週間に1回投与の有効性を評価する。
- ハイゼントラの週1回投与から切り替えた2週間に1回投与における用量を評価する。
- ハイゼントラを2週間に1回投与した被験者の生活の質(QOL)を評価する。
[PK substudy主要目的]
ハイゼントラの週1回投与及び2週間に1回投与の薬物動態特性を評価する。
試験デザイン
多施設共同、1群クロスオーバー、非盲検、プロスペクティブ
対象
3ヵ月以上安定したIgG補充療法を受けているPID患者又はSID患者※25例
投与方法
2つの期間(パート1、パート2)で構成され、在宅で最長15ヵ月間皮下投与する。
パート1:ハイゼントラ0.1~0.2g(100~200mg)/kg体重を週1回
パート2:ハイゼントラ0.2~0.4g(200~400mg)/kg体重(パート1で投与された1回量の2倍量)を2週間に1回(パート1の最終投与から2週間後にパート2の初回投与を実施)
投与期間
パート1:12週間
パート2:最長52週間
主要評価項目
ハイゼントラの2週間に1 回投与期間中の局所有害事象及びすべての有害事象の年間発現回数
副次評価項目
ハイゼントラの週1回投与期間中及び2週間に1回投与期間中の被験者あたりの感染症の年間発現回数、ハイゼントラの週1回投与期間中及び2週間に1回投与期間中の血清中IgGトラフ値、QOL尺度(SF-36、CHQ-PF28及びCHQ-CF87)
PK substudy 主要評価項目
ハイゼントラの週1回投与及び2週間に1回投与の定常状態での薬物動態パラメータ、dAUC(週1回投与は7日間、2週間に1回投与は14日間算出)、最高血清中IgG濃度、最高血清中IgG濃度到達時間
解析計画
主要評価項目及び副次評価項目の正式な統計的仮説検定は計画しなかった。統計解析及び結論は、試験データの要約統計量に基づいた。
ハイゼントラの2週間に1回投与の安全性、忍容性及び有効性の評価、ハイゼントラの週1回投与から切り替えた2週間に1回投与の評価及びQOL評価は、被験者データの一覧表のレビュー及び要約統計量に基づき実施した。
[安全性]
解析対象集団は、ATS(All Treated Subjects)Analysis Setとした。要約には有害事象及びバイタルサインを含めた。週1回投与、2週間に1回投与、及び両投与レジメンを併合した集団について、ハイゼントラ投与期間の局所有害事象及び全有害事象の年間発現回数を被験者別及び投与レジメン別に要約統計量を算出した。
[有効性]
解析対象集団は、ATS Analysis Set及びPP(Per Protocol)Analysis Setとした。
感染症の年間発現回数は、被験者別及び投与レジメン別に要約統計量を算出した。
[QOL]
解析対象集団は、 mITT(Modified Intent-to-Treat)Analysis Setとした。
SF-36は、サマリースコア(身体的側面、精神的側面)及び健康領域スケール(身体機能、日常役割機能[身体]、体の痛み、全体的健康感、活力、社会生活機能、日常役割機能[精神]及び心の健康)の結果の要約統計量をTスコア(0~100点)に基づき算出し、それぞれのベースラインからの経時変化を示した。
CHQ-PF28及びCHQ-CF87は、サマリースコアの結果の要約統計量を投与レジメン別に標準化されたスコアに基づき算出し、ベースラインからの経時変化を示した。10歳未満の小児にはCHQ-PF28、10歳以上の小児にはCHQ-CF87を用い、解析は別々に行った。
[薬物動態]
血清中薬物動態濃度データは、要約統計量を用いて投与レジメン別に要約した。要約統計量には被験者数、算術平均値、標準偏差(SD)、中央値、最小値、最大値に加えて幾何平均値及び変動係数(CV)を含めた。
週1回投与及び2週間に1回投与から得られた血清中IgGの濃度-時間データから、血清中IgG濃度についてノンコンパートメント解析を実施した。
PKパラメータの解析には、実際の検体採取時刻(ハイゼントラの投与開始時点からの実際の経過時間)、実際の投与に要した時間及び実際の投与量を用いた。
血清中薬物動態パラメータ(AUC0-tau、dAUC、Ctrough、Cmax、Tmax、Cmin、CLss)は、濃度データと同じ要約統計量を用いて投与レジメン別に記述的に要約した。Tmaxについては、被験者数、中央値、最小値及び最大値のみ算出した。
週1回投与と2週間に1回投与のdAUCを幾何平均比(GMR)及びその90%信頼区間(CI)に基づき比較した。また、週1回投与と2週間に1回投与の用量で補正していないCmax、Cmin及びCtroughをGMR及びその90%CIに基づき比較した。
※ PID及びSID患者を対象としていたが、SID患者は組み入れられなかった。
PID:原発性免疫不全症候群、SID:続発性免疫不全症候群、PK:薬物動態、SF-36:Short-Form 36、CHQ-PF28:Child Health Questionnaire Parent Form 28、
CHQ-CF87:Child Health Questionnaire Child Form 87、dAUC:投与量で補正したAUC0-tau、
ATS Analysis Set:登録された被験者のうち、投与レジメンにかかわらず本剤が1回以上投与されたすべての被験者
投与方法

患者背景
| ATS Analysis Set (N=25) | ||||
| 年齢(歳) | 平均値(標準偏差) | 23.6(17.93) | ||
| 中央値(範囲) | 16.0(6~66) | |||
| 年齢層、n(%) | 12歳未満 | 8(32.0) | ||
| 12歳以上16歳未満 | 3(12.0) | |||
| 16歳以上18歳未満 | 4(16.0) | |||
| 18歳以上 | 10(40.0) | |||
| 性別、n(%) | 男性 | 14(56.0) | ||
| 女性 | 11(44.0) | |||
| 人種、n(%) | 白人 | 24(96.0) | ||
| その他: 父親がアフリカ人、 母親が白人 | 1(4.0) | |||
| 体重(kg) | 平均値(標準偏差) | 57.28(23.369) | ||
| 中央値(範囲) | 64.00(19.0~96.0) | |||
| BMI(kg/m2) | 平均値(標準偏差) | 22.41(6.089) | ||
| 中央値(範囲) | 21.77(13.7~37.0) | |||
| 疾患の内訳 | 分類不能型 免疫不全症 (CVID) | n(%) | 13(52.0) | |
| 診断データあり、n | 13 | |||
| 期間、年 | 平均値(標準偏差) | 4.5(3.99) | ||
| 中央値(範囲) | 2.4(1~12) | |||
| 重症複合 免疫不全症 (SCID) | n(%) | 2(8.0) | ||
| 診断データあり、n | 0 | |||
| 期間、年 | 平均値(標準偏差) | - | ||
| 中央値(範囲) | - | |||
| その他の 免疫不全症 | n(%) | 10(40.0) | ||
| 診断データあり、n | 5 | |||
| 期間、年 | 平均値(標準偏差) | 3.2(4.30) | ||
| 中央値(範囲) | 1.5(1~11) | |||
ハイゼントラの投与状況
| パート1 (週1回投与) (N=25) | パート2 (2週間に1回投与) (N=24) | ||
| 試験期間 | 日数 | 2,414 | 6,878 |
| 投与回数 | 309 | 476 | |
| 注射部位数 | 平均値 (SD) | 2.5 (0.77) | 4.1 (0.79) |
| 中央値 (範囲) | 2 (1~5) | 4 (2~8) | |
| 注射部位あたりの用量 (mL) | 平均値 (SD) | 12.8 (5.19) | 14.5 (5.47) |
| 中央値 (範囲) | 11.5 (5~27) | 15.0 (3~37) | |
| 注入時間 (時間) | 平均値 (SD) | 0.94 (0.677) | 1.23 (0.564) |
| 中央値 (範囲) | 0.75 (0.3~0.9) | 1.09 (0.4~2.5) | |
| 被験者あたりの用量 (mg/kg体重) | 平均値 (SD) | 112.14 (32.453) | 218.62 (64.118) |
| 中央値 (範囲) | 109.25 (52.0~239.7) | 211.36 (113.0~473.0) | |
N:解析対象集団の被験者数
n:被験者数
忍容性及び安全性
海外第Ⅲ相試験において、週1回投与群と2週間に1回投与群における有効性・安全性に差異はなく、IgGの全体的な曝露量も同程度であったと考えられました。
主要評価項目:2週間に1回投与期間中の局所有害事象及びすべての有害事象の年間発現回数
(ATS Analysis Set)
| パート2(2週間に1回投与) | 有害事象 (N=24) | 副作用 (N=24) |
| すべて | 22(91.7) | 3(12.5) |
| 重篤 | 1(4.2) | 0 |
| 試験中止に至った | 1(4.2) | 1(4.2) |
| 死亡 | 0 | 0 |
| 局所 | 3(12.5) | - |
| 被験者あたりの年間発現回数、件a) | 0.18(0.509) | - |
重篤な有害事象は、入院を要する胃腸炎1例、試験中止に至った有害事象は、片頭痛1例であった。
a)平均値(標準偏差)
ATS:All Treated Subjects
海外第Ⅲ相試験における有害事象及び副作用
本試験に組み入れられ、ハイゼントラの皮下投与を受けたすべての被験者[パート1(週1回投与) 25例、パート2(2週間に1回投与) 24例、期間全体25例]で評価しました。
有害事象発現率は、パート1(週1回投与)80.0%(20/25例)、パート2(2週間に1回投与)91.7%(22/24例)でした。
副作用の発現率は、パート1(週1回投与)16.0%(4/25例)、パート2(2週間に1回投与)12.5%(3/24例)でした。
副作用の内訳は、パート1(週1回投与)で疲労、注射部位内出血、注射部位疼痛、錯感覚が各4.0%(1/25例)、パート2(2週間に1回投与)で注射部位紅斑、注射部位腫瘤、片頭痛が各4.2%(1/24例)でした。
重篤な有害事象は、パート1(週1回投与)では認められず、パート2(2週間に1回投与)で1例(入院を要する胃腸炎)に認められました。
投与中止に至った有害事象は、パート1(週1回投与)では認められず、パート2(2週間に1回投与)で1例(片頭痛)に認められました。
試験期間中に死亡例は認められませんでした。
有効性
副次評価項目:感染症の年間発現回数(ATS Analysis Set)
| パート1(週1回投与) (N=25) | パート2(2週間に1回投与) (N=24) | 試験期間全体 (N=25) | ||
| 感染症の発現例数、n(%) | 12(48.0) | 13(54.2) | 18(72.0) | |
| 感染症の発現件数、件 | 16 | 17 | 33 | |
| 試験期間、日a) | 2,414 | 6,878 | 9,268 | |
| 感染症の年間発現回数、回/人年b) | 2.42 | 0.90 | 1.30 | |
| 被験者あたりの 感染症の年間発現 回数、回/人年 | 平均値(標準偏差) | 2.43(3.146) | 1.22(1.641) | 1.41(1.501) |
| 中央値(範囲) | 0(0~11.91) | 1.02(0~5.14) | 0.83(0~6.23) | |
b)年間発現回数=発現回数/試験期間日数×365.25
ATS:All Treated Subjects
感染症の発現リスクは季節によって変わるうえ、本試験のパート1(週1回投与)とパート2(2週間に1回投与)では観察期間が異なり、認められた感染症の発現回数に影響する可能性があるため、年換算した有効性の結果をパート1とパート2で一律に比較することはできない。
薬物動態
血清IgG濃度(IgPro20_4005試験)3)
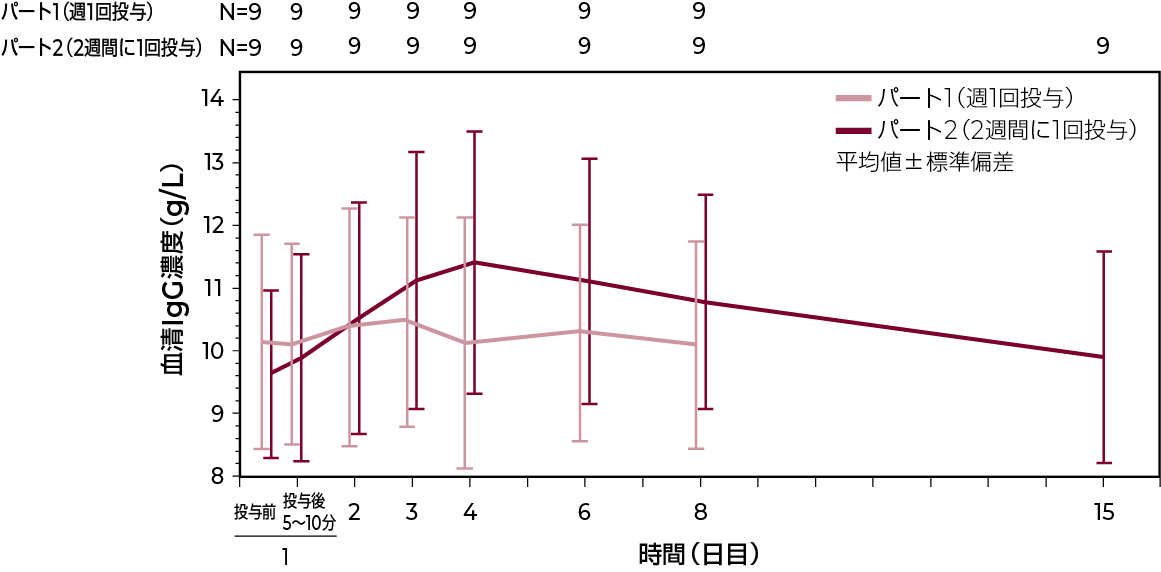
原発性免疫不全症候群患者を対象とした海外第Ⅲ相試験において、ハイゼントラを週1回及び2週間に1回クロスオーバー投与した結果、本剤の血清IgG濃度パラメータ(Cmax、トラフ濃度及びdAUC)は両投与間で類似していました。
血清中IgG濃度-時間プロファイル
薬物動態パラメータ
| パラメータ | パート1 (週1回投与) (N=9) | パート2 (2週間に1回投与) (N=9) |
| 平均投与量 (mg/kg体重) | 109±17.2 | 207±38.4 |
| Cmax(g/L) | 10.63±1.70 | 11.63±2.03 |
| Ctrough(g/L) | 10.04±1.63 | 9.86±1.68 |
| AUC0-tau (h*g/L) | 1,707±294 | 3,561±594 |
| dAUC (h*g/L/mg) | 0.24±0.04 | 0.26±0.04 |
AUC0-tau:週1回投与はAUC0-7日、2週間に1回投与はAUC0-14日、
dAUC:投与量で補正したAUC0-tau
薬物動態プロファイルの比較(母集団薬物動態解析※による予測値 vs 血清中IgG濃度-時間プロファイルの実測値)
6. 用法及び用量(抜粋)
〈無又は低ガンマグロブリン血症〉
通常、人免疫グロブリンGとして50~200mg(0.25~1mL)/kg体重を週1回皮下投与する。2週間に1回投与する場合には、1週あたりの用量の2倍量(100~400mg(0.5~2mL)/kg体重)を皮下投与する。なお、患者の状態に応じて、1週もしくは2週あたりの投与量及び投与回数は適宜増減する。
7. 用法及び用量に関連する注意
〈効能共通〉
7.1 皮下注射にのみ使用すること。静脈内に投与してはならない。
7.2 本剤の投与開始にあたっては、医療施設において、必ず医師によるか、医師の直接の監督のもとで投与を行うこと。本剤による治療開始後、医師により適用が妥当と判断された患者については、自己投与も可能である。[8.4参照]
〈無又は低ガンマグロブリン血症〉
7.3 静注用人免疫グロブリン製剤から本剤に切り換える患者において、本剤の1週あたりの投与量は、静注用人免疫グロブリン製剤を3週間間隔で投与していた場合はその1/3量、また、4週間間隔で投与していた場合はその1/4量から開始し、初回投与は静注用人免疫グロブリン製剤の最終投与1週間後に投与すること。2週間に1回投与する場合には1週あたりの2倍量とすること。以降の本剤の投与量は、感染頻度や重症度など本剤による治療の臨床反応及び血清IgG濃度を参考に調節すること。
7.4 人免疫グロブリン製剤による治療歴のない患者を対象とした本剤の臨床試験は実施されていない。人免疫グロブリン製剤による治療歴のない患者に対して本剤による導入を行う場合は、感染頻度や重症度など本剤による治療の臨床反応と血清IgG濃度を参考に、投与量を慎重に調節すること。また、1週もしくは2週あたりの投与量を数日に分割して投与するなど、投与間隔の調節も考慮すること。
14. 適用上の注意(抜粋)
14.2 薬剤投与時の注意
14.2.2 本剤は腹部、大腿部、上腕部、腰部側面等に皮下投与すること。投与量に応じて複数箇所からの投与を検討し、投与部位は少なくとも5cm離すこと。
14.2.4 投与速度
(1)部位あたりの投与量は、初回投与では20mL以下とし、以降の投与では患者の状態に応じて最大50mLまで増量することができる。投与速度は、初回投与では部位あたり20mL/時間以下とし、患者の状態に応じて最大50mL/時間まで徐々に増加することができる。
(2)注射部位反応が報告されているので、推奨投与速度を守り、投与毎に投与部位を変えること。
「禁忌を含む注意事項等情報」等については、電子化された製品添付文書をご参照ください。