本試験で用いられた用量の一部には本邦において承認されていないものを含みます。本試験では用量別の投与群が設定されておらず、承認用量を超える投与群(601mg/kg体重から800mg/kg体重)を除外することが困難であること、併せて原著との不整合が生じることを防ぐため、承認外の用量を含む内容を記載しました。
海外第Ⅲ相試験:ZLB03_002CR試験(海外データ)
小児および成人の原発性免疫不全症候群におけるピリヴィジェンの有効性および安全性の検討
社内資料:臨床概要(2020年2月21日承認)(ZLB03_002CR試験)(承認時評価資料)
試験概要
目的
原発性免疫不全症候群患者(小児および成人)におけるピリヴィジェンの有効性、安全性を評価する。
試験デザイン
前向き、多施設共同、単群、非盲検、第Ⅲ相試験
対象
原発性液性免疫不全(X連鎖無ガンマグロブリン血症または分類不能型免疫不全症)と診断され、6ヵ月間以上静注用人免疫グロブリン(IVIG)補充療法を行う患者80例※1
- 3~70歳の男女
- 直近6ヵ月間以上、IVIG補充療法を3週または4週間隔で投与されている
- 組み入れ前6ヵ月以内のIgGトラフ値が1回以上4g/L以上となっている
投与方法
ピリヴィジェン200~800mg/kg体重を、3週または4週間隔で12ヵ月間にわたって静脈内投与した。
[用量および投与間隔]
本試験前のIVIG補充療法と同一の用量および投与間隔を、試験期間中にわたって維持
[投与速度]
0.5mg/kg体重/分で開始、投与後最初の30分間の忍容性が良好であれば次の30分間は1.0mg/kg体重/分に上昇可、この場合も忍容性が良好であればさらなる上昇も可とした。最大投与速度は、1~3回目の投与では4.0mg/kg体重/分、4回目以降の投与では8.0mg/kg体重/分とした。
評価項目
【有効性主要評価項目】
ITTSにおける患者あたりの急性重篤細菌感染症(肺炎、菌血症/敗血症、骨髄炎/化膿性関節炎、細菌性髄膜炎、内臓膿瘍)の年間発現回数
【有効性副次評価項目】
PPSにおける患者あたりの急性重篤細菌感染症の年間発現回数など
【安全性主要評価項目】
ピリヴィジェン投与と時間的に関連のある有害事象、ピリヴィジェンとの因果関係が否定できない時間的に関連のある有害事象、投与ごとおよび患者ごとのすべての有害事象の発現率、重症度および因果関係など
解析計画
[有効性主要/副次評価項目]
解析対象集団は、ITTSおよびPPSとした。患者あたりの急性重篤細菌感染症の年間発現回数に関しては、ヒストリカルデータ※2を用いた基準値より、片側97.5%CIの上限値が1.0回/人・年以上になるという帰無仮説を検定した。急性重篤細菌感染症の発現回数は、ポアソン分布となると仮定した。
[安全性評価項目]解析対象集団はSDSとしたが、ITTSと同一の集団であった。有害事象は国際医薬用語集(MedDRA)Version9.0を用いてコード化し、器官別大分類および基本語別に解析した。時間的に関連のある有害事象は投与中または投与終了後48時間以内に発現した有害事象と定義し、ピリヴィジェンとの因果関係は4段階(関連なし、関連があるかもしれない、おそらく関連あり、関連あり)で評価した。
※1:治験実施計画50例以上を予定
※2:FDAより定められたIgG切り替えの有効性指標
CI:confidence interva(l 信頼区間)、ITTS:intention-to-treat se(t 治療意図に基づく解析対象集団)、PKAS:pharmacokinetic analysis se(t 薬物動態解析対象集団)、PPKAS:per-protocol pharmacokinetic analysis set(治験実施計画書に適合した薬物動態解析対象集団)、PPS:per-protocol set(治験実施計画書に適合した解析対象集団)、SAS:safety analysis set(安全性解析対象集団:ピリヴィジェンを1回以上投与された被験者集団)、SDS:safety data set(安全性解析対象集団:ピリヴィジェンを1回以上投与された被験者集団)
有効性
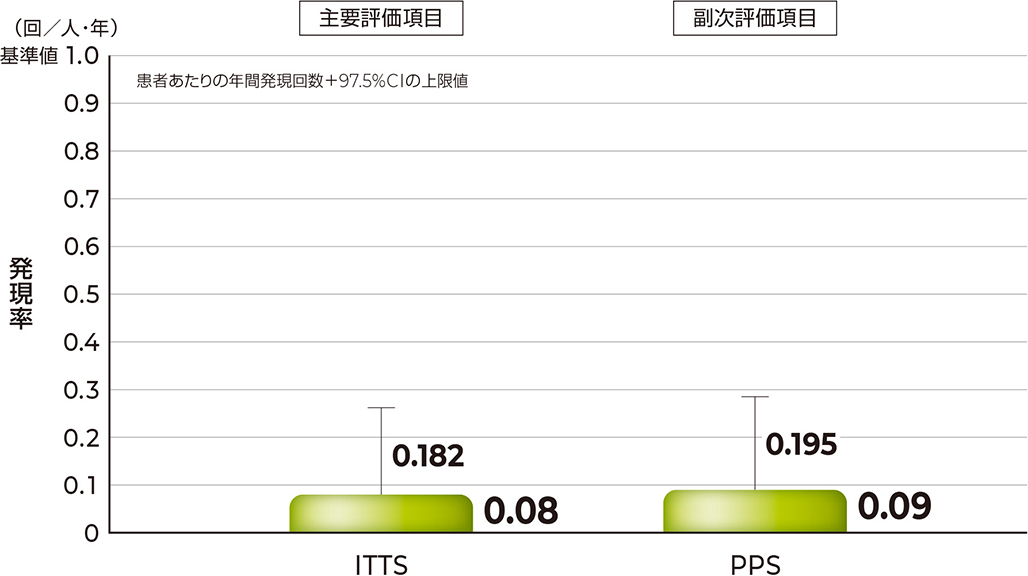
ITTSにおける患者あたりの急性重篤細菌感染症の年間発現回数および片側97.5%CIの上限値は、それぞれ0.08および0.182回/人・年であり、この上限値は事前に設定した基準値1.0回/人・年を下回りました。
急性重篤細菌感染症が発現した被験者の割合(48時間以内)(ITTSおよびPPS)
| 集団 | N | 発現例数(%) | 発現回数 | 合計試験参加 日数(日) | 患者あたりの年間発現 回数(回/人・年) | 片側97.5%CIの 上限値 |
| 〔主要評価項目〕 急性重篤細菌感染症(ITTS) | 80 | 6(7.5) | 6 | 26,198 | 0.08 | 0.182 |
| 肺炎 | 80 | 3(3.8) | 3 | 26,198 | 0.04 | ー |
| 化膿性関節炎a) | 80 | 1(1.3) | 1 | 26,198 | 0.01 | ー |
| 骨髄炎b) | 80 | 1(1.3) | 1 | 26,198 | 0.01 | ー |
| 内臓膿瘍c) | 80 | 1(1.3) | 1 | 26,198 | 0.01 | ー |
| 〔副次評価項目〕 急性重篤細菌感染症(PPS) | 70 | 6(8.6) | 6 | 24,463 | 0.09 | 0.195 |
| 肺炎 | 70 | 3(4.3) | 3 | 24,463 | 0.04 | ー |
| 化膿性関節炎a) | 70 | 1(1.4) | 1 | 24,463 | 0.01 | ー |
| 骨髄炎b) | 70 | 1(1.4) | 1 | 24,463 | 0.01 | ー |
| 内臓膿瘍c) | 70 | 1(1.4) | 1 | 24,463 | 0.01 | ー |
a)基本語:細菌性関節炎、b)基本語:感染、c)基本語:後腹膜膿瘍(いずれもMedDRA Version 9.0)
急性重篤細菌感染症の患者あたりの年間発現回数(ITTSおよびPPS)
安全性
有害事象
有害事象発現率は97.5%(78/80例)であり、30%以上の頻度で頭痛67.5%(54/80例)、咳嗽33.8%(27/80例)、副鼻腔炎31.3%(25/80例)が認められました。
ピリヴィジェンとの因果関係が否定できない有害事象※の発現率は41.3%(33/80例)であり、10%以上の頻度で頭痛30.0%(24/80例)、悪心12.5%(10/80例)、悪寒および疲労各11.3%(9/80例)がみられました。
重篤な有害事象が16例(死亡例1例を含む)に38件認められ、そのうち1例で発現した5件の重篤な有害事象(過敏症、悪寒、疲労、浮動性めまい、体温上昇)はピリヴィジェンと関連ありと判定されました。主な重篤な有害事象として、肺炎が3例(3件)にみられました。
有害事象による投与中止は4例(死亡例1例を含む)で報告されました。死亡例を除く3例のうち1例はピリヴィジェンと関連ありと判定された5件の重篤な有害事象、別の1例は悪寒(ピリヴィジェンとの因果関係:関連あり[以下、因果関係のみを示す])、頭痛(おそらく関連あり)、上気道感染(関連なし)および鼻炎(関連なし)、残り1例は嘔吐(おそらく関連あり)により投与中止に至りました。
多臓器不全による死亡が1例に認められましたが、ピリヴィジェンとの関連は否定されました。
※ ピリヴィジェンと「関連あり」、「おそらく関連あり」または「関連があるかもしれない」と治験責任医師が判断した有害事象
| 基本語 | (N=80) |
| ピリヴィジェンとの因果関係が否定できない有害事象 | 33(41.3) |
| 頭痛 | 24(30.0) |
| 悪心 | 10(12.5) |
| 悪寒 | 9(11.3) |
| 疲労 | 9(11.3) |
| 嘔吐 | 6(7.5) |
| 疼痛 | 5(6.3) |
| 背部痛 | 4(5.0) |
| 発熱 | 4(5.0) |
| インフルエンザ様疾患 | 3(3.8) |
| 呼吸困難 | 2(2.5) |
| 下痢 | 2(2.5) |
| 振戦 | 2(2.5) |
| 下腹部痛 | 1(1.3) |
| 腹部圧痛 | 1(1.3) |
| 関節痛 | 1(1.3) |
| 無感情 | 1(1.3) |
| 無力症 | 1(1.3) |
| 血圧低下 | 1(1.3) |
| 体温上昇 | 1(1.3) |
| 気管支炎 | 1(1.3) |
| 基本語 | (N=80) |
| 胸痛 | 1(1.3) |
| 冷汗 | 1(1.3) |
| クームス試験陽性 | 1(1.3) |
| 浮動性めまい | 1(1.3) |
| 紅斑 | 1(1.3) |
| 顔面痛 | 1(1.3) |
| びくびく感 | 1(1.3) |
| 全身症状 | 1(1.3) |
| 過敏症 | 1(1.3) |
| インフルエンザ | 1(1.3) |
| 注入部位疼痛 | 1(1.3) |
| 注射部位疼痛 | 1(1.3) |
| 白血球減少症 | 1(1.3) |
| 筋骨格硬直 | 1(1.3) |
| 鼻咽頭炎 | 1(1.3) |
| 頚部痛 | 1(1.3) |
| 末梢性浮腫 | 1(1.3) |
| 四肢痛 | 1(1.3) |
| 咽喉頭疼痛 | 1(1.3) |
| 処置による高血圧 | 1(1.3) |
| そう痒症 | 1(1.3) |
| 胃不快感 | 1(1.3) |
n:患者数
MedDRA Version 9.0
n(%)
※ピリヴィジェンと「関連あり」、「おそらく関連あり」または「関連があるかもしれない」と治験責任医師が判断した有害事象
本試験で用いられた用量の一部には本邦において承認されていないものを含みます。本試験では用量別の投与群が設定されておらず、承認用量を超える投与群(601mg/kg体重から800mg/kg体重)を除外することが困難であること、併せて原著との不整合が生じることを防ぐため、承認外の用量を含む内容を記載しました。
海外第Ⅲ相継続試験:ZLB05_006CR試験(海外データ)
小児および成人の原発性免疫不全症候群におけるピリヴィジェンの安全性、有効性ならびに最大投与速度の忍容性の検討
社内資料:臨床概要(2020年2月21日承認)(ZLB03_002CR試験)(承認時評価資料)
試験概要
目的
原発性免疫不全症候群患者(小児および成人)におけるピリヴィジェンの安全性、有効性ならびに最大投与速度(12.0mg/kg体重/分[7.2mL/kg体重/時間])の忍容性を評価する。
試験デザイン
前向き、多施設共同、単群、非盲検、第Ⅲ相試験
対象
原発性液性免疫不全(X連鎖無ガンマグロブリン血症または分類不能型免疫不全症)と診断された患者55例。うち45例は、12ヵ月間のZLB03_002CR試験を完了し本試験に移行した継続被験者、残り10例は、本試験前に6ヵ月間以上静注用人免疫グロブリン(IVIG)補充療法(200~800mg/kg体重)を3週または4週間隔で実施した新規被験者
投与方法
ピリヴィジェン200~800mg/kg体重を、3週または4週間隔で静脈内投与した。投与期間はピリヴィジェンの米国での上市まで、または他の海外第Ⅲ相試験への切り替えまでとした。
[用量および投与間隔]
本試験前に用いた最終3回の用量と同量ならびに同じ投与間隔を、試験期間中にわたって維持
[投与速度]
低速で開始、投与後最初の30分間の忍容性が良好であれば投与速度の上昇を可とした。継続被験者に関しては、ZLB03_002CR試験における最大投与速度(8.0mg/kg体重/分)の忍容性が良好であれば、12.0mg/kg体重/分までの上昇も可とした。新規被験者の最大投与速度は、4.0mg/kg体重/分とした。
評価項目
[有効性評価項目]患者あたりの急性重篤細菌感染症(肺炎、菌血症/敗血症、骨髄炎/化膿性関節炎、細菌性髄膜炎、内臓膿瘍)の年間発現回数など
[安全性評価項目]ピリヴィジェン投与と時間的に関連のある有害事象、ピリヴィジェンとの因果関係が否定できない時間的に関連のある有害事象、投与ごとおよび患者ごとのすべての有害事象の発現率、重症度および因果関係、最大投与速度に対する忍容性など
解析計画
[有効性評価項目]解析対象集団は、ITTSとした。
[安全性評価項目]解析対象集団はSDSとしたが、ITTSと同一の集団であった。有害事象は国際医薬用語集(MedDRA)Version11.0を用いてコード化し、器官別大分類および基本語別に解析した。時間的に関連のある有害事象は投与中または投与終了後48時間または72時間以内に発現した有害事象と定義し、ピリヴィジェンとの因果関係は4段階(関連なし、関連があるかもしれない、おそらく関連あり、関連あり)で評価した。投与ごとの最大投与速度(8.0mg/kg体重/分以下[低速投与]および8.0mg/kg体重/分超[高速投与])について解析した。最大投与速度は四捨五入し、整数で表記した。
有効性
患者あたりの急性重篤細菌感染症の年間発現回数(ITTS)[有効性評価項目]
試験期間中に55例中1例(1.8%)で認められた肺炎1件が急性重篤細菌感染症と判定されました。ITTSにおける患者あたりの急性重篤細菌感染症の年間発現回数は0.018回/人・年でした。
安全性
有害事象(SDSおよびITTS)
有害事象発現率は94.5%(52/55例)であり、20%以上の頻度で頭痛38.2%(21/55例)、副鼻腔炎25.5%(14/55例)、咳嗽21.8%(12/55例)、悪心20.0%(11/55例)が認められました。
ピリヴィジェンとの因果関係が否定できない有害事象※の発現率は43.6%(24/55例)であり、5%以上の頻度で頭痛29.1%(16/55例)、発熱、疲労、疼痛、悪寒、蕁麻疹各5.5%(3/55例)がみられました。
重篤な有害事象が11例に17件認められ、いずれもピリヴィジェンとの因果関係が否定されました。17件のうち2件は同一被験者1例に発現した小腸閉塞で、残り15件の事象はすべて異なっていました。
ピリヴィジェンとの因果関係が否定できない有害事象の蕁麻疹による投与中止が1例で報告されました。死亡例は認められませんでした。
※ピリヴィジェンと「関連あり」、「おそらく関連あり」または「関連があるかもしれない」と治験責任医師が判断した有害事象
最大投与速度に対する忍容性(SDS)
継続被験者のSDSにおいて、最大投与速度がピリヴィジェン投与と時間的に関連のある有害事象(72時間以内)の発現に及ぼす影響を検討しました。
対象となったのは継続被験者のSDSにおけるピリヴィジェンの総投与回数690回から投与速度不明の2回を除いた688回であり、うち423回は低速投与群(投与速度8.0mg/kg体重/分以下)、265回は高速投与群(投与速度8.0mg/kg体重/分超)に分類されました。
時間的に関連のある有害事象の発現率を検討するため、各群で総投与回数に占める時間的に関連のある有害事象が発現した投与回数の割合を算出したところ、低速投与群では0.362、高速投与群では0.113でした。また、ピリヴィジェンとの因果関係が否定できない時間的に関連のある有害事象が認められた投与回数は、低速投与群で108回、高速投与群で2回であり、その発現率はそれぞれ0.255、0.008でした。
国内第Ⅲ相試験:IgPro10_3004試験
小児および成人の日本人原発性免疫不全症候群におけるピリヴィジェンの薬物動態と安全性の検討
社内資料:臨床概要(2020年2月21日承認)(IgPro10_3004試験)(承認時評価資料)
試験概要
目的
日本人原発性免疫不全症候群患者(小児および成人)におけるピリヴィジェンの薬物動態および安全性を検討する。
試験デザイン
前向き、多施設共同、単群、非盲検、第Ⅲ相試験
対象
原発性免疫不全症候群(X連鎖無ガンマグロブリン血症または分類不能型免疫不全症など)と診断され、6ヵ月間以上静注用人免疫グロブリン(IVIG)補充療法を行う日本人患者11例
- 同意取得時に6歳以上かつ19kg以上の男女
- 直近6ヵ月間のIVIG補充療法を一定用量で3週または4週間隔で定期的に投与されている
- 組み入れ前6ヵ月以内のIgGトラフ値が1回以上5g/L以上となっている
投与方法
12週間のウォッシュイン/ウォッシュアウト期間後に、ピリヴィジェン200~600mg/kg体重を、3週または4週間隔で最長4ヵ月間にわたって静脈内投与した。
[用量および投与間隔]
体重/分で開始、投与後最初の30分間の忍容性が良好であれば、個々の患者の状態をふまえつつ治験責任医師の裁量で慎重に投与速度を上げた。最大投与速度は、1回目の投与では2.0mg/kg体重/分、2回目4.0mg/kg体重/分、3回目8.0mg/kg体重/分、4回目12.0mg/kg体重/分、5回目12.0mg/kg体重/分であった。
評価項目
[主要評価項目]血清IgG濃度に基づく薬物動態パラメータ
[有効性評価項目]初回投与前および最終投与前の血清IgGトラフ値
[安全性評価項目]有害事象の発現率、有害事象の重症度など
解析計画
[主要評価項目]解析対象集団は、PKASおよびPPKASとした。ノンコンパートメント解析を用いて、薬物動態パラメータの幾何平均および95%CIを算出した。
[有効性評価項目]解析対象集団は、SASとした。記述統計量(患者数、最小値、最大値、中央値、平均値および標準偏差)を用いて解析した。
[安全性評価項目]解析対象集団は、有効性評価項目と同様とした。ピリヴィジェンの初回投与日またはそれ以降に発現した有害事象は、国際医薬用語集(MedDRA)Version 21.0を用いてコード化した。
薬物動態
ピリヴィジェンの静脈内投与により得られた血清IgGの最高血清濃度(Cmax)の幾何平均は、3週間隔投与群で16.4g/L、4週間隔投与群で13.4g/Lであり、いずれも投与開始後約1時間で到達しました。
用量で補正した血清IgG濃度に基づく薬物動態パラメータ推定値(PKAS/PPKAS)[主要評価項目]
| パラメータ(単位) | 3週間隔投与群(N=2) | 4週間隔投与群(N=8) | |
| Cmax(g/L) | 幾何平均(95%CI) | 16.4(NR) | 13.4(9.98~18.0) |
| Cmin(g/L) | 幾何平均(95%CI) | 10.3(NR) | 7.98(5.90~10.8) |
| Tmax(h) | 中央値(範囲) | 1.19(0.92~1.47) | 1.14(0.62~23.37) |
| AUC0-last(g・h/L) | 幾何平均(95%CI) | 5,891(NR) | 6,239(4,713~8,258) |
| dAUC(g・h/L)a) | 幾何平均(95%CI) | 0.402(NR) | 0.424(0.302~0.594) |
| CL(mL/h) | 幾何平均(95%CI) | 2.49(NR) | 2.36(1.68~3.31) |
NR:2例のため算出せず
a)mgあたりの用量で補正したAUC
社内資料:臨床概要(2020年2月21日承認)(IgPro10_3004試験)(承認時評価資料)
社内資料:臨床薬理試験(2020年2月21日承認)(IgPro10_3004試験)(承認時評価資料)
有効性
ピリヴィジェンの最終投与前の血清IgGトラフ値の平均値±標準偏差は、3週間隔投与群で10.0g/L、4週間隔投与群で8.0±3.8g/Lでした。
血清IgGトラフ値(SAS)[有効性評価項目]
| 3週間隔投与群(N=2) | 4週間隔投与群(N=9) | 全被験者(N=11) | |
| 初回投与前濃度(g/L) | |||
| n 平均値±標準偏差 中央値 (範囲) | 2 10.9±2.0 10.9 (9.47~12.33) | 9 8.6±4.0 7.5 (5.09~17.81) | 11 9.0±3.8 8.1 (5.09~17.81) |
| 最終投与前濃度(g/L) | |||
| n 平均値±標準偏差 中央値 (範囲) | 2 10.0±2.5 10.0 (8.26~11.83) | 8 8.0±3.8 6.9 (5.37~16.82) | 10 8.4±3.5 7.5 (5.37~16.82) |
| 変化量a)(g/L) | |||
| n 平均値±標準偏差 中央値 (範囲) | 2 -0.9±0.5 -0.9 (-1.21~-0.50) | 8 -0.3±0.4 -0.3 (-0.99~0.48) | 10 -0.4±0.5 -0.4 (-1.21~0.48) |
血清総IgG濃度の推移(PKAS/PPKAS)
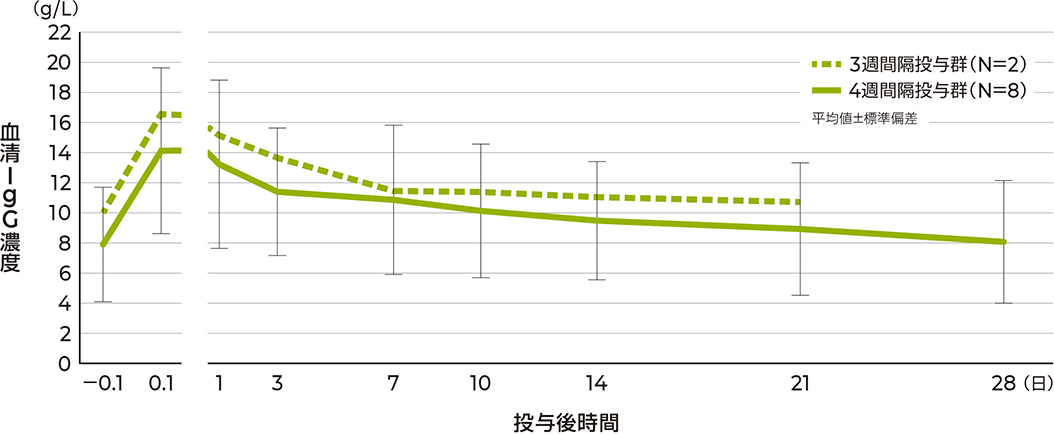
注)初回投与前60~1分および投与後3~20分は異なるスケールで表示した。
3週間隔投与群は2例のため標準偏差は表示していない。
社内資料:臨床概要(2020年2月21日承認)(IgPro10_3004試験)(承認時評価資料)
社内資料:臨床薬理試験(2020年2月21日承認)(IgPro10_3004試験)(承認時評価資料)
安全性
曝露量(SAS)
10例が試験を完了し、総投与期間の平均値(範囲)は14.9(4.1~16.3)週間でした。9例が8.0mg/kg体重/分以上の投与速度に忍容性を示し、うち5例が12.0mg/kg体重/分(7.2mL/kg体重/時間)の投与速度でのピリヴィジェン投与に忍容性を示しました。
有害事象(SAS)
有害事象発現率は72.7%(8/11例)で、ピリヴィジェンと関連のある有害事象は1例(9.1%)、治療後72時間以内に発現した時間的に関連のある有害事象は3例(27.3%)に認められました。基本語別の有害事象は、上咽頭炎27.3%(3/11例)のほか、上気道感染、乾癬、発疹、蕁麻疹、残存乳歯、上腹部痛、下痢、節足動物咬傷、転倒、皮膚擦過傷、注入部位不快感、発熱、上室性頻脈、C-反応性蛋白増加がそれぞれ9.1%(1/11例)認められました。重篤な有害事象および死亡に至った有害事象はみられませんでした。投与中止および試験中止に至った有害事象として乾癬が1例報告されましたが、ピリヴィジェンとの関連は認められませんでした。
| (N=11、I=43) | |||
| n(%) | E | AERI | |
| 有害事象 ピリヴィジェンと関連のない ピリヴィジェンと関連のある 時間的に関連のある(治療後72時間以内) ピリヴィジェンと関連のある、または時間的に関連のある(治療後72時間以内) | 8(72.7) 8(72.7) 1(9.1) 3(27.3) 3(27.3) | 19 18 1 3 3 | 0.442 0.419 0.023 0.070 0.070 |
| 有害事象の重症度 軽度 中等度 重度 | 5(45.5) 3(27.3) 0 | 12 4 | 0.279 0.093 |
| 重篤な有害事象 | 0 | ||
| 有害事象による投与中止 ピリヴィジェンと関連のない ピリヴィジェンと関連のある | 1(9.1) 1(9.1) 0 | 1 1 | 0.023 0.023 |
| 有害事象による試験中止 ピリヴィジェンと関連のない ピリヴィジェンと関連のある | 1(9.1) 1(9.1) 0 | 1 1 | 0.023 0.023 |
社内資料:臨床概要(2020年2月21日承認)(IgPro10_3004試験)(承認時評価資料)
6. 用法及び用量(抜粋)
〈無又は低ガンマグロブリン血症〉
通常、1回人免疫グロブリンGとして200~600mg(2~6mL)/kg体重を3~4週間隔で点滴静注又は緩徐に静注する。患者の状態によって適宜増減する。
14. 適用上の注意(抜粋)
14.2 薬剤投与時の注意
14.2.2 投与速度
〈効能共通〉
(1)ショック等の副作用は初日の投与開始30分以内、また投与速度を上げた際に起こる可能性があるので、これらの時間帯については特に注意すること。
[7.1、9.7.1参照]
〈無又は低ガンマグロブリン血症〉
(2)初回の投与開始から約30分は0.6mL/kg体重/時間で投与し、副作用等の異常所見が認められなければ、投与速度を7.2mL/kg体重/時間まで徐々に上げることができる。その後の投与は、耐容した速度で開始することができる。
「禁忌を含む注意事項等情報」等については、電子化された製品添付文書をご参照ください。