海外第Ⅲ相試験:PRIMA試験(海外データ)1)、 2)
PRIMA試験では、CIDPの急性期治療および維持療法としてのピリヴィジェンの有効性と安全性を検討しました。
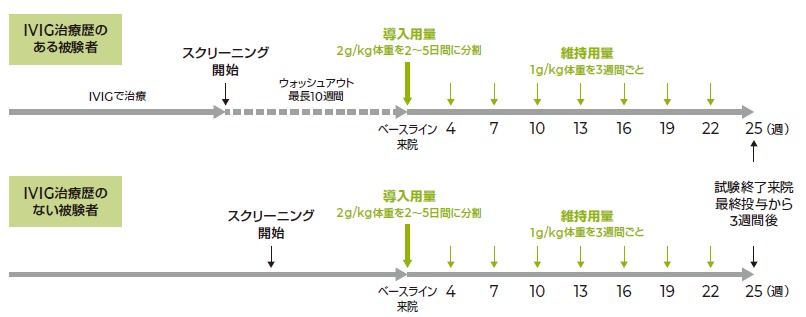
試験概要
目的
CIDP患者におけるピリヴィジェンの有効性および安全性を検討する。
対象
ニューロパチーに起因する2肢以上での運動神経障害および感覚神経障害、または対称性の運動神経障害のみが進行性もしくは再発性に認められ、CIDPと診断された患者28例
試験デザイン
多施設共同、単群、非盲検、第Ⅲ相試験(検証試験)
方法
IVIG治療歴のある被験者には最長10週間のウォッシュアウト期間を設け、ウォッシュアウト期間中に調整INCATスコアで1ポイント以上悪化した被験者に対して、ピリヴィジェンの投与を開始した。IVIG治療歴のない被験者はスクリーニング後にピリヴィジェンの投与を開始した。いずれの被験者にも下記用量でピリヴィジェンを投与した。
[急性期治療:導入用量]
2g/kg体重を連続する2~5日間に分割して静脈内投与した。1日最大用量は1g/kg体重とした。
[維持療法:維持用量]
1g/kg体重を1日または連続する2日間に分割して3週間ごとに7回(4、7、10、13、16、19および22週時)静脈内投与した。
評価項目
[主要評価項目]
調整INCATスコアでのレスポンダー※1率
[副次評価項目]
調整INCATスコアの1ポイント以上の改善、またはMRC合計スコアの3ポイント以上の改善のいずれかによる初回反応までの時間、調整INCATスコア、最大握力(マーチン握力計を使用)およびMRC合計スコアのベースラインからの変化量
[安全性評価項目]
有害事象、臨床検査値、血清IgG濃度、バイタルサインなど
解析計画
[主要評価項目]
解析対象集団はFASとした。単群のレスポンダー率の点推定値およびその両側95%Wilsonスコア信頼区間(CI)の下限値の解析に基づき実施した。ピリヴィジェン群のレスポンダー率の両側95%WilsonスコアCIの下限値が35%※2を上回る場合に、本試験は成功とみなした。
[副次評価項目]
解析対象集団はFASとした。調整INCATスコア、最大握力およびMRC合計スコアのベースラインからの変化量は、平均値およびパラメトリックCI、並びにノンパラメトリックホッジス・レーマン点推定値およびCIを算出した。さらに、調整INCATスコアおよびMRC合計スコアでの初回反応までの時間は、Kaplan-Meier法に基づき算出した。
[サブグループ解析]
IVIG治療歴のある被験者とない被験者における探索的なサブグループ解析を実施した。
[安全性評価項目]
解析対象集団はFASとした※3。有害事象、臨床検査値、血清IgG濃度、バイタルサインなどを記述的に解析した。
※1 レスポンダー:調整INCATスコア(0~10ポイント)において、1ポイント以上改善した被験者をレスポンダーとした。
※2 CIDP治療にIVIGを用いたプラセボ対照比較試験(ICE試験)3)から得たヒストリカルプラセボ群におけるレスポンダ-率の95%CIの上限値に基づき設定した。
※3 安全性評価項目の解析対象集団はSDSとしたが、SDSはFASと同一の集団であった。
INCATスコア
INCATスコアは、上肢と下肢の機能を0(正常)~10(上肢または下肢での目的を持った動きが不可能)で評価する尺度である。
調整INCATスコアとは、上肢機能の0(正常)から1(軽微な症状)または1から0への変化は、臨床的に意義がないとの判断により、悪化または改善として記録しないスコアのことである。
1)社内資料:臨床概要(臨床的有効性)(PRIMA試験)(承認時評価資料)
2)Léger JM, et al.:J Peripher Nerv Syst. 2013;18(2):130-140 [https://creativecommons.org/licenses/by/3.0/]
利益相反:本試験はCSLベーリングより資金助成を受けた。本論文の著者のうち4人はCSLベーリングの社員である。
3)Hughes RA, et al.:Lancet Neurol. 2008;7(2):136-144
CIDP:chronic inflammatory demyelinating polyneuropathy(慢性炎症性脱髄性多発根神経炎)、EFNS:European federation of neurological societies(欧州神経学会)、FAS:full analysis set(最大の解析対象集団)、INCAT:inflammatory neuropathy cause and treatment、IVIG:intravenous immunoglobulin(静注用人免疫グロブリン)、ITTS:intention-to-treat set(intention-to-treat解析対象集団)、LOCF:last observation carried forward、MRC:Medical research council(英国医学研究審議会)、PNS:Peripheral nerve society(末梢神経学会)、PPS:per-protocol set(治験実施計画書に適合した解析対象集団)、PSDS:pre-randomization safety data set(無作為化前安全性解析対象集団)、RSDS:rescue medication safety data set(救済薬安全性解析対象集団)、R-ODS:rasch-built overall disability scale、SDS:safety data set(安全性解析対象集団)
患者背景
| FAS (N=28) | ||
| 年齢、歳 | 平均値(標準偏差) | 58.7(14.34) |
| 中央値(範囲) | 58.0(22~79) | |
| 性別、n(%) | 男性 | 18(64.3) |
| 女性 | 10(35.7) | |
| 人種、n(%) | 白人 | 28(100) |
| 体重、kg | 平均値(標準偏差) | 82.3(16.72) |
| 中央値(範囲) | 83.0(50~118) | |
| BMI、kg/m2 | 平均値(標準偏差) | 27.1(4.38) |
| 中央値(範囲) | 27.9(18~36) | |
| 身長、cm | 平均値(標準偏差) | 173.9(10.27) |
| 中央値(範囲) | 172.0(158~195) | |
| CIDPと診断されてからの期間、n(%) | 1年以下 | 9(32.1) |
| 1年超、2年以下 | 4(14.3) | |
| 2年超、10年以下 | 12(42.9) | |
| 10年超 | 3(10.7) | |
有効性
試験終了来院時のレスポンダー率は約60%でした。
主要評価項目である試験終了来院時のレスポンダー率[両側95%WilsonスコアCI]は60.7%[42.41~76.43]であり、両側95%WilsonスコアCIの下限値が35%※1を上回ったことから、本試験は成功とみなされました。
調整INCATスコアでのレスポンダー率 主要評価項目 主要評価項目のサブグループ解析
試験終了来院時の調整INCATスコアでのレスポンダー率(FAS)
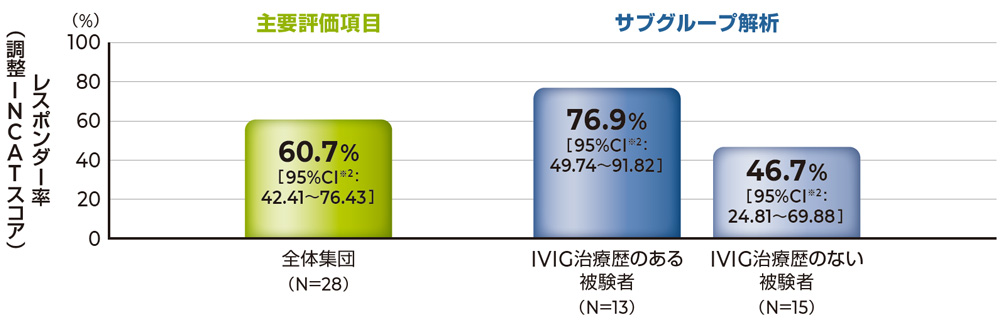
注 試験終了来院時の欠測値はLOCF法により補完
※1 ICE試験のヒストリカルプラセボ群におけるレスポンダー率の95%CIの上限値に基づき、レスポンダー率の両側95%WilsonスコアCIの下限値が35%を上回る場合に、本試験は成功とみなした
※2 両側95%WilsonスコアCI
1)社内資料:臨床概要(臨床的有効性)(PRIMA試験)(承認時評価資料)
2)Léger JM, et al.:J Peripher Nerv Syst. 2013;18(2): 130-140 [https://creativecommons.org/licenses/by/3.0/]より作成
ピリヴィジェン投与後4週時に約30%、7週時に50%の患者が治療への反応を示しました。
調整INCATスコアでの初回反応までの時間 副次評価項目 副次評価項目のサブグループ解析
Kaplan-Meier法に基づく調整INCATスコアでの初回反応までの時間(FAS)
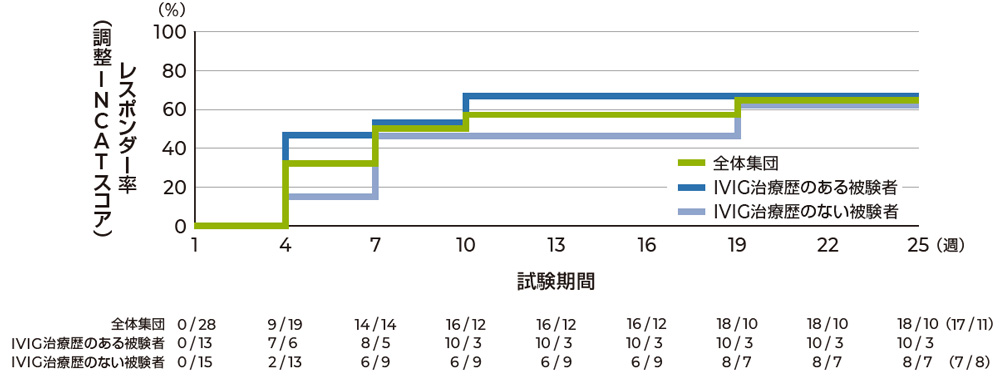
注 各週における「観察時点までに少なくとも1回は反応を示した被験者数/1回も反応を示さなかった被験者数」を表した。なお、19および22週時には反応を示したが、25週時に反応を示さなかった被験者が1例いたため、25週時点で反応を示した被験者の数と示さなかった被験者の数を( )内に表した。
2)Léger JM, et al.:J Peripher Nerv Syst. 2013;18(2):130-140 [https://creativecommons.org/licenses/by/3.0/]
安全性
副作用(FAS)
全投与期間における有害事象発現率は78.6%(22/28例)、副作用発現率は60.7%(17/28例)でした。
治療期間別の副作用
急性期治療期間における副作用発現率は46.4%(13/28例)でした。5%以上に発現した副作用は、頭痛25.0%(7/28例)、無力症および高血圧各10.7%(3/28例)、インフルエンザ様疾患、悪心および溶血各7.1%(2/28例)でした。
維持療法期間における副作用発現率は39.3%(11/28例)でした。5%以上に発現した副作用は、頭痛14.3%(4/28例)、高血圧および白血球減少症各7.1%(2/28例)でした。
急性期治療期間の副作用発現状況(FAS)
| 副作用発現状況(N=28) | |
| 副作用 | 13(46.4) |
| 重篤な副作用 | 2(7.1)※ |
| 投与中止に至った副作用 | 2(7.1)※ |
| 死亡例 | 0 |
| 5%以上に発現した副作用 | |
| 頭痛 | 7(25.0) |
| 無力症 | 3(10.7) |
| 高血圧 | 3(10.7) |
| インフルエンザ様疾患 | 2(7.1) |
| 悪心 | 2(7.1) |
| 溶血 | 2(7.1) |
維持療法期間の副作用発現状況(FAS)
| 副作用発現状況(N=28) | |
| 副作用 | 11(39.3) |
| 重篤な副作用 | 0 |
| 投与中止に至った副作用 | 0 |
| 死亡例 | 0 |
| 5%以上に発現した副作用 | |
| 頭痛 | 4(14.3) |
| 高血圧 | 2(7.1) |
| 白血球減少症 | 2(7.1) |
1)社内資料:臨床概要(臨床的有効性)(PRIMA試験)(承認時評価資料)
ピリヴィジェンの【用法及び用量】
- 慢性炎症性脱髄性多発根神経炎の筋力低下の改善
通常、成人には1日に人免疫グロブリンGとして400mg(4mL)/kg体重を5日間連日点滴静注する - 慢性炎症性脱髄性多発根神経炎の運動機能低下の進行抑制(筋力低下の改善が認められた場合)
通常、成人には人免疫グロブリンGとして「1,000mg(10mL)/kg体重を1日」又は「500mg(5mL)/kg体重を2日間連日」を3週間隔で点滴静注する。
製造工程はほぼ同一であるものの、タンパク質濃度と投与方法が異なる2つのIgG製剤[ピリヴィジェン10%静注(本剤)とハイゼントラ20%皮下注]を同時に開発し、同一の試験(PATH試験)をもって製造販売承認を同時申請したため、紹介するデータには一部承認外の成績が含まれます。
PATH試験におけるハイゼントラの臨床成績はこちらをご参照ください。
国際共同第Ⅲ相試験:PATH試験(日本人を含む海外データ)4~7)
PATH試験では、IVIGへの依存性が確認されたCIDP患者に対する急性期治療および維持療法としてのピリヴィジェンの有効性と安全性を検討しました。
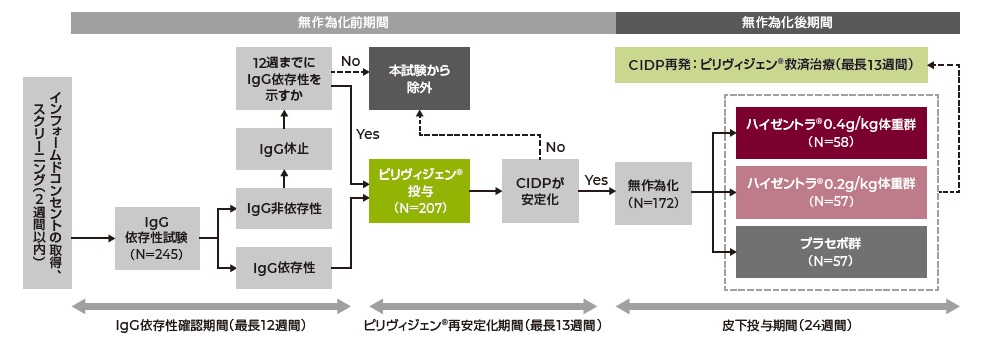
6)より改変
試験概要
目的
CIDPの維持療法において異なる2用量のハイゼントラ(0.2g/kg体重または0.4g/kg体重)の有効性および安全性をプラセボと比較評価する。
対象
EFNS/PNS診断基準2010により、definite CIDPまたはprobable CIDPと診断され、組み入れ前8週間以内にIVIGの投与を受けた患者207例(日本人患者15例を含む)
試験デザイン
多施設共同、並行群間、二重盲検、無作為化、プラセボ対照、第Ⅲ相試験(検証試験)
方法
2つの期間(無作為化前期間、無作為化後期間)で構成され、ピリヴィジェンの有効性および安全性は無作為化前期間中のピリヴィジェン再安定化期間、および無作為化後期間中のピリヴィジェン救済治療期間に検討。
[無作為化前期間]
IgG依存性確認期間およびピリヴィジェン再安定化期間で構成された。
- IgG依存性確認期間(最長12週間):2週間のスクリーニング期間後、IgGの継続投与が必要である被験者を組み入れるために、IgG依存性を確認(IgG依存性試験:IgG投与後IgGの効果が減弱した際、被験者がCIDPの臨床的悪化を示すか否かを判断)する期間を設けた。
- ピリヴィジェン再安定化期間(最長13週間):IgG依存性確認期間中にCIDPが増悪※1した被験者に対し、急性期治療(導入用量)としてピリヴィジェン2g/kg体重を2~5日間(日本:連続する5日間)に分割して静脈内投与した。その後、維持療法(維持用量)として、ピリヴィジェン1g/kg体重を1日または連続する2日間に分割して3週間ごとに3回または4回静脈内投与した。
ピリヴィジェン再安定化期間の最後の連続する2回の来院で、INCATスコアが安定している(ただしINCATスコアはスクリーニング時より悪化していない)被験者を無作為化後期間に移行した。
[無作為化後期間]
ハイゼントラの有効性および安全性を検討する皮下投与期間(最長24週間)。
IgGへの依存性が確認されCIDPの状態が安定した被験者を組み入れ、ハイゼントラ0.2g/kg体重群、0.4g/kg体重群、またはプラセボ群に無作為に割り付け、週1回投与を2回の投与セッションに分けて皮下投与を行った。これを1日または連続する2日間に分割し24週間投与した。
皮下投与期間中にCIDPが再発※2した被験者には、ハイゼントラまたはプラセボの投与を中止し、救済治療としてピリヴィジェンを投与するピリヴィジェン救済治療期間(最長13週間)を設けた。ピリヴィジェン救済治療期間には、ピリヴィジェン2g/kg体重を1回のみ静脈内投与、もしくはその後に維持用量としてピリヴィジェン1g/kg体重を3週間間隔で最大4回静脈内投与した。
評価項目
[主要評価項目]
皮下投与期間中にCIDPの再発が認められた、またはその他の理由により試験(皮下投与)を中止した被験者の割合
[有効性評価項目]
INCATスコア、R-ODSパーセンタイルスコア、平均握力(マーチン握力計を使用)、MRC合計スコア、調整INCATスコアで最初の改善が認められるまでの時間
[安全性評価項目]
曝露量、有害事象、臨床検査値、バイタルサイン、身体検査および心電図(日本のみ)
解析計画
[主要評価項目]
解析対象集団は、ITTSおよびPPSとした。主要評価項目において3群間の傾向を検討するため、第1種過誤率を0.025(片側)としたCochran-Armitage傾向検定を実施した。優越性傾向が示された場合は、片側Fisher確率検定を用いて、各群の対比較を実施した。また、CIDPの再発以外の理由で試験を中止した被験者を含めることによって生じる潜在的バイアスを検討するために感度分析を行った。
[有効性評価項目]
再安定化期間における解析対象集団はPSDS、救済治療期間における解析対象集団はRSDSとした。記述統計を用いて試験来院別に要約した。調整INCATスコアでのレスポンダー率について、日本人サブグループ解析を実施した。
[安全性評価項目]
解析対象集団は有効性評価項目と同様とした。曝露量、有害事象、臨床検査値、バイタルサイン、身体所見および心電図(日本のみ)を要約した。
※1 CIDPの増悪:IgG依存性確認期間中に以下の基準に該当した場合。
- 治験実施計画書の改訂3以前:調整INCATスコアの1ポイント以上の増加
- 治験実施計画書の改訂3以降:調整INCATスコアの1ポイント以上の増加、R-ODS総スコアの4ポイント以上の減少、または平均握力(片手)の8kPa以上の低下
※2 CIDPの再発:ベースライン(ピリヴィジェン再安定化期間の試験終了来院時)と比較して、INCATスコアが1ポイント以上増加した場合(上肢スコアが0から1に増加したINCATスコアの1ポイント増加は除く)、または上肢スコアが1から0に減少し、下肢スコアが1ポイント増加したためINCATスコアが不変の場合。
4)社内資料:臨床概要(臨床的有効性)(PATH試験)(承認時評価資料)
5)van Schaik IN, et al.:Lancet Neurol. 2018;17(1):35-46
利益相反:本試験はCSLベーリングより資金助成を受けた。著者にCSLベーリングよりコンサルタント料、謝礼、研究費を受領している者が含まれる。本論文の著者のうち4人はCSLベーリングの社員である。
6)van Schaik IN, et al.:Trials. 2016;17(1):345 [https://creativecommons.org/licenses/by/4.0/]
利益相反:本試験はCSLベーリングより資金助成を受けた。著者にCSLベーリングよりコンサルタント料、謝礼、研究費を受領している者が含まれる。本論文の著者のうち2人はCSLベーリングの社員である。
7)Mielke O, et al.:J Peripher Nerv Syst. 2019;24(1):72-79
利益相反:本試験はCSLベーリングより資金助成を受けた。著者にCSLベーリングよりコンサルタント料、謝礼、研究費を受領している者が含まれる。本論文の著者のうち4人はCSLベーリングの社員である。
ピリヴィジェンの【用法及び用量】
- 慢性炎症性脱髄性多発根神経炎の筋力低下の改善
通常、成人には1日に人免疫グロブリンGとして400mg(4mL)/kg体重を5日間連日点滴静注する。 - 慢性炎症性脱髄性多発根神経炎の運動機能低下の進行抑制(筋力低下の改善が認められた場合)
通常、成人には人免疫グロブリンGとして「1,000mg(10mL)/kg体重を1日」又は「500mg(5mL)/kg体重を2日間連日」を3週間隔で点滴静注する。
ハイゼントラの【用法及び用量】(抜粋)
- 慢性炎症性脱髄性多発根神経炎の運動機能低下の進行抑制(筋力低下の改善が認められた場合)
通常、成人には人免疫グロブリンGとして1週あたり200mg(1mL)/kg体重を1日又は連続する2日で分割して皮下投与するが、患者の状態に応じて、最大400mg(2mL)/kg体重から投与を開始することもできる。なお、維持用量は200〜400mg/kg体重で適宜増減する。
患者背景
| 再安定化期間 | 救済治療期間 | ||
| PSDS (N=207) | RSDS (N=60) | ||
| 年齢、歳 | 平均値(標準偏差) | 56.5(12.76) | 53.0(11.84) |
| 中央値(範囲) | 58.2(24.7~82.7) | 53.3(24.7~77.2) | |
| 年齢層、n(%) | 18歳以上、65歳以下 | 150(72.5) | 49(81.7) |
| 65歳超 | 57(27.5) | 11(18.3) | |
| 性別、n(%) | 男性 | 131(63.3) | 40(66.7) |
| 女性 | 76(36.7) | 20(33.3) | |
| 人種、n(%) | 白人 | 186(89.9) | 54(90.0) |
| アジア人 | 17(8.2) | 5(8.3) | |
| うち日本人 | 15(7.2) | 5(8.3) | |
| その他 | 4(1.9) | 1(1.7) | |
| 体重a)、kg | 平均値(標準偏差) | 82.2(18.33) | 82.6(18.08) |
| 中央値(範囲) | 82.0(41.8~133.0) | 81.8(41.7~130.2) | |
| BMIa、b)、kg/m2 | 平均値(標準偏差) | 27.3(4.99) | 26.9(4.59) |
| 中央値(範囲) | 26.9(17.6~49.4) | 26.2(18.4~41.6) | |
| 初めてCIDPと診断されてからの 期間、年c) | 平均値(標準偏差) | 4.7(5.19) | 4.0(4.31) |
| 中央値(範囲) | 3.0(0.1~33.5) | 2.66(0.2~22.2) | |
| スクリーニング時のEFNS/PNS CIDP診断基準、n(%) | Definite | 185(89.4) | 56(93.3) |
| Probable | 22(10.6) | 4(6.7) | |
| スクリーニング/ベースライン時の INCATスコアd)、ポイント | 平均値(標準偏差) | 2.7(1.67) | 1.8(1.47) |
| 中央値(範囲) | 3.0(0~8) | 2.0(0~7) | |
BMI算出被験者数=205例(PSDS)、および60例(RSDS)
c)CIDPの初回診断からの時間(年)=(同意取得日-初回診断日+1)/365.25
d)ピリヴィジェン 再安定化期間のスクリーニング時および皮下投与期間のベースライン時のINCATスコア
有効性
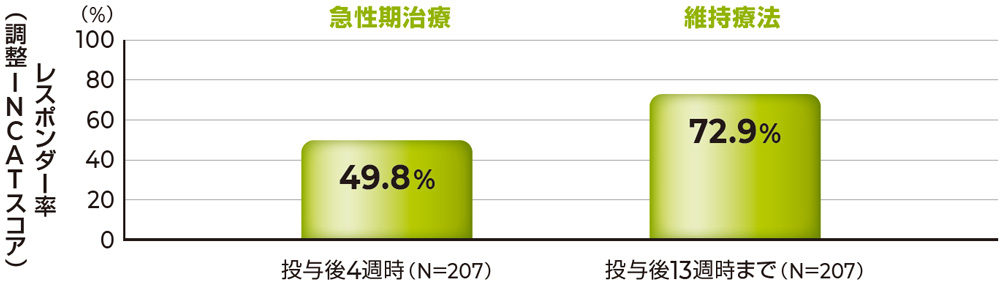
ピリヴィジェン再安定化期間におけるレスポンダー率は、投与後4週時で約50%、維持療法期間である投与後13週時までで約70%でした。
再安定化期間の有効性[無作為化前ピリヴィジェン投与] 有効性評価項目
投与後4週時および13週時までのレスポンダー率(PSDS)
再安定化期間の有効性パラメータのまとめ(PSDS)
| 再安定化期間 | ||||
| n | 急性期治療 (投与後4週時) | n | 維持療法 (投与後10週時) | |
| ベースラインからの変化量(平均値±標準偏差) | ||||
| INCATスコア、ポイント | 199 | -0.9±1.10 | 190 | -1.2±1.12 |
| R-ODSパーセンタイルスコア、ポイント | 83 | 5.1±9.78 | 146 | 5.7±13.45 |
| 利き手の握力、kPa | 102 | 8.0±13.89 | 177 | 12.2±16.18 |
| MRC合計スコア、ポイント | 102 | 2.8±4.27 | 178 | 3.6±4.34 |
| 規定の有効性評価項目※1のいずれかで 改善が認められた被験者、%(n) | 207 | 90.8(188)※2 | ||
※2 規定の有効性評価項目のいずれかで改善が認められた被験者の割合は、投与後10週時ではなく、再安定化期間中(投与後13週時まで)にいずれかの改善が認められた被験者の割合を示す。
4)社内資料:臨床概要(臨床的有効性)(PATH試験)(承認時評価資料)
7)Mielke O, et al.:J Peripher Nerv Syst. 2019;24(1):72-79
皮下投与期間の有効性[無作為化後ハイゼントラ投与](ITTS) 主要評価項目
(検証的解析結果)
皮下投与期間中にCIDPの再発、またはその他の理由により試験を中止した被験者の割合は、ハイゼントラ0.2g/kg体重群38.6%(22/57例)、0.4g/kg体重群32.8%(19/58例)であり、プラセボ群の63.2%(36/57例)と比較して有意に低く(それぞれp=0.007およびp<0.001、片側Fisher確率検定)、ハイゼントラ0.2g/kg体重群および0.4g/kg体重群のプラセボ群に対する優越性が検証されました。
4)社内資料:臨床概要(臨床的有効性)(PATH試験)(承認時評価資料)
5)van Schaik IN, et al.:Lancet Neurol. 2018;17(1):35-46
安全性
再安定化期間の副作用[無作為化前ピリヴィジェン投与]
再安定化期間における有害事象発現率は48.3%(100/207例)、副作用発現率は27.5%(57/207例)でした。
急性期治療期間における副作用発現率は20.3%(42/207例)でした。2%以上に発現した副作用は、頭痛9.2%(19/207例)、悪心2.9%(6/207例)でした。
維持療法期間における副作用発現率は13.9%(28/201例)でした。2%以上に発現した副作用は、頭痛3.5%(7/201例)でした。
再安定化期間の副作用発現状況(PSDS)
| 副作用発現状況 | ||
| 急性期治療期間(N=207) | 維持療法期間(N=201) | |
| 副作用 | 42(20.3) | 28(13.9) |
| 重篤な副作用 | 2(1.0)※1 | 5(2.5)※2 |
| 投与中止に至った副作用 | 0 | 4(2.0)※3 |
| 死亡例 | 0 | 0 |
| いずれかの期間で2%以上に発現した副作用 | ||
| 頭痛 | 19(9.2) | 7(3.5) |
| 悪心 | 6(2.9) | 2(1.0) |
※1 片頭痛および発疹が各1例に発現
※2 拡張期血圧上昇、CIDP、過敏症、肺塞栓症および呼吸不全が各1例に発現
※3 肺塞栓症、腎不全、呼吸不全および頭痛が各1例に発現
4)社内資料:臨床概要(臨床的有効性)(PATH試験)(承認時評価資料)
7)Mielke O, et al.:J Peripher Nerv Syst. 2019;24(1):72-79
救済治療期間の副作用[無作為化後ピリヴィジェン投与]
救済治療期間における有害事象発現率は28.3%(17/60例)、副作用発現率は15.0%(9/60例)でした。2%以上に発現した副作用は、頭痛6.7%(4/60例)、悪心5.0%(3/60例)および溶血3.3%(2/60例)でした。
救済治療期間の副作用発現状況(RSDS)
| 副作用発現状況(N=60) | |
| 副作用 | 9(15.0) |
| 重篤な副作用 | 0 |
| 投与中止に至った副作用 | 0 |
| 死亡例 | 0 |
| 2%以上に発現した副作用 | |
| 頭痛 | 4(6.7) |
| 悪心 | 3(5.0) |
| 溶血 | 2(3.3) |
※ PATH試験では治験実施計画書の改訂があり、改訂3以前に組み入れた被験者にはピリヴィジェン救済治療期間において、急性期治療(導入用量として2g/kg体重のピリヴィジェンを1回投与)のみを実施した。改訂3以降に組み入れた被験者には、急性期治療の後に維持療法(1g/kg体重のピリヴィジェンを3週間ごとに最長9週間投与)を実施した。そのため、ピリヴィジェン救済治療期間の安全性成績では、急性期治療と維持療法を分けず、救済治療期間全体として記載した。
4)社内資料:臨床概要(臨床的有効性)(PATH試験)(承認時評価資料)
ピリヴィジェンの【用法及び用量】
- 慢性炎症性脱髄性多発根神経炎の筋力低下の改善
通常、成人には1日に人免疫グロブリンGとして400mg(4mL)/kg体重を5日間連日点滴静注する。 - 慢性炎症性脱髄性多発根神経炎の運動機能低下の進行抑制(筋力低下の改善が認められた場合)
通常、成人には人免疫グロブリンGとして「1,000mg(10mL)/kg体重を1日」又は「500mg(5mL)/kg体重を2日間連日」を3週間隔で点滴静注する。
ハイゼントラの【用法及び用量】(抜粋)
- 慢性炎症性脱髄性多発根神経炎の運動機能低下の進行抑制(筋力低下の改善が認められた場合)
通常、成人には人免疫グロブリンGとして1週あたり200mg(1mL)/kg体重を1日又は連続する2日で分割して皮下投与するが、患者の状態に応じて、最大400mg(2mL)/kg体重から投与を開始することもできる。なお、維持用量は200〜400mg/kg体重で適宜増減する。
「禁忌を含む注意事項等情報等」については、電子添文をご参照ください。
ハイゼントラの「禁忌を含む注意事項等情報等」については、電子添文をご参照ください。