開発の経緯
血友病A[血液凝固第VIII因子(factor VIII:FVIII)欠乏症]は、X染色体連鎖劣性遺伝性の出血性疾患である。
血友病A治療は、FVIII製剤[血漿由来FVIII又は遺伝子組換えFVIII(rFVIII)]の投与によりFVIIIレベルを上昇させ、FVIIIの欠乏状態を一時的に補正することで、出血エピソードの管理(オンデマンド療法)又は出血エピソードの抑制(定期補充療法)を主な目的としている。
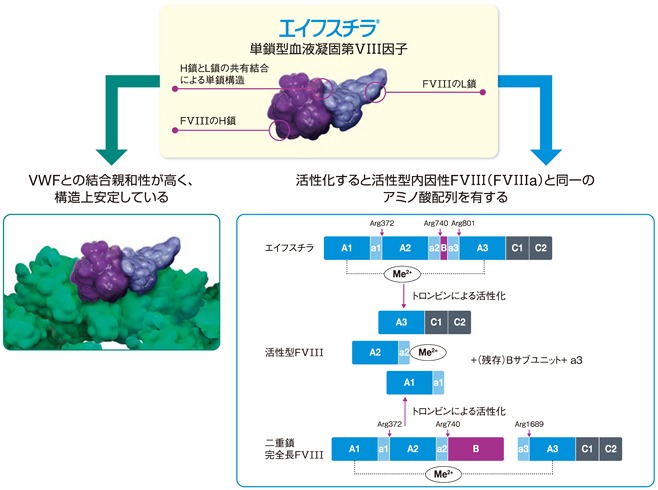
エイフスチラ静注用[一般名:ロノクトコグ アルファ(遺伝子組換え)]は、CSLベーリング社が薬物動態(PK)特性を改善し、投与頻度の少ない(週2回投与)高純度製剤を血友病A患者に提供することを目的として開発した遺伝子組換え単鎖FVIII(recombinant, single-chain coagulation FVIII:単鎖rFVIII)である。エイフスチラは、内因性FVIIIのBドメインの大部分及び隣接する酸性a3ドメインの4アミノ酸が取り除かれているため、Bドメインとa3ドメイン間のfurin切断部位で切断されず、単鎖FVIII分子として発現する。
エイフスチラは、天然の担体タンパク質であるフォン・ヴィレブランド因子(VWF)に対して、他のrFVIII分子よりも強力な親和性を示す。VWFはFVIIIに結合し、FVIII分子を初期のタンパク質分解から保護することから、エイフスチラではより強力な親和性によりPKプロファイルが改善するものと考えられる。
エイフスチラの製造及び調製段階において、ヒト又は動物由来のタンパク質は添加しておらず、2つの独立したウイルス除去工程を含んでおり、感染因子の伝播リスクの軽減を図っている。
エイフスチラの臨床開発プログラムは、成人及び小児の血友病A患者(FVIII活性1%未満)を対象に、PKプロファイル、安全性及び有効性を検討することを目的としており、国際共同第I/III相試験(1001試験)及び海外第III相試験(3002試験)の完了した2試験から構成され、日本人への投与は1001試験において実施された。
その結果、良好なPKプロファイルが示されるとともに、出血エピソードに対するオンデマンド療法、定期補充療法及び周術期の補充療法(外科手術時の補充療法)において、安全性と有効性が示されたことから、「血液凝固第VIII因子欠乏患者における出血傾向の抑制」を効能又は効果として2017年9月に承認を取得した。
なお、米国では2016年5月に、EUでは2017年1月に承認を取得した。
JPN-AFS-1855
2024年6月改訂
製品特徴
- フォン・ヴィレブランド因子との結合親和性が高く、構造上安定した単鎖型の遺伝子組換え血液凝固第VIII因子(単鎖rFVIII)製剤である。
- 血中半減期は14.2時間、AUC0-infは1,910 IU・hr/dLであった※1。
- 50 IU/kg、週3回の投与で血漿中FVIII活性を4.4%に維持できる可能性が示唆された※2。
- 出血エピソードに対する治療成功率*は92.3%であり、93.5%が1回又は2回投与で止血可能であった※1。
*治験責任医師の総合的臨床評価で「著効」又は「有効」であった患者の割合
- 小児患者(12歳未満)において出血エピソードに対する治療成功率*は96.3%であり、95.7%が1回又は2回投与で止血可能であった※3。
*治験責任医師の総合的臨床評価で「著効」又は「有効」であった患者の割合
- 週2回又は3回投与の定期補充療法による自然出血の年間出血回数の中央値は0件/人年であった※1、3。
- 周術期止血管理に対し、治験責任医師が評価した止血効果は、すべての手術で「著効」又は「有効」であった※1。
- 重大な副作用として、ショック、アナフィラキシー(いずれも頻度不明)があらわれることがある。主な副作用として呼吸困難、浮動性めまい、錯感覚、発疹、紅斑、そう痒症、発熱、注射部位疼痛、悪寒、熱感(1%未満)、インヒビターの発現(頻度不明)が報告されている(詳細については、電子添文の副作用及び臨床成績の安全性の結果を参照)。
※1:12~65歳の重症(FVIII活性<1%)血友病A患者を対象とした国際多施設共同非盲検第I/III相試験
※2:12歳以上の血友病A患者における母集団PK解析に基づくシミュレーション
※3:12歳未満の重症(FVIII活性<1%)血友病A患者を対象とした海外多施設共同非盲検第III相試験
JPN-AFS-1855
2024年6月改訂
作用機序
血液凝固第VIII因子の役割とエイフスチラの作用点
エイフスチラ(一般名:ロノクトコグ アルファ)は、遺伝子組換え単鎖FVIII製剤であり、生体由来の完全長FVIIIのBドメインの大部分、及び隣接する酸性a3ドメインの4アミノ酸残基(完全長FVIIIのアミノ酸番号765~1652番)を取り除いている。重鎖(H鎖)と軽鎖(L鎖)の間には新しい配列が挿入され、そこには新規のN結合型糖鎖付加部位が組み込まれている。生体由来完全長FVIIIのBドメインとa3ドメインの間に存在するfurin切断部位が除去されているため、単鎖FVIII分子として発現される。
エイフスチラは活性化されると内因性の完全長FVIIIから生じる活性型FVIII(FVIIIa)と同一のアミノ酸配列を有するため、FVIII欠乏患者に対し、FVIIIを一時的に補正することで、出血傾向を改善する。
なお、単鎖構造であるため、フォン・ヴィレブランド因子(VWF)※との高い親和性が得られる1)ことから薬物動態プロファイルの改善が期待できる。
※フォン・ヴィレブランド因子(VWF):FVIIIの担体タンパク質。FVIIIと結合して活性のない安定した複合体を形成し、血漿中に存在する。FVIIIを初期のタンパク質分解から保護するとともに、止血の際には血管損傷部位でコラーゲンと結合し、一次止血に寄与する。
エイフスチラの構造と作用機序
1)Zollner S et al.: Thromb Res. 2014; 134(1): 125-131
本研究はCSLベーリングの支援により行われた。著者はCSLベーリングの社員である。
JPN-AFS-1855
2024年6月改訂
薬物動態
1.血漿中濃度
(1)単回投与後の薬物動態評価(日本人及び外国人データ)1)
12~65歳の血友病A患者(FVIII活性<1%)を対象とし、エイフスチラ50 IU/kg単回投与後の薬物動態パラメータを下表に示す。
日本人で報告された平均値は全て全体で報告された値の範囲内であった。
エイフスチラ50 IU/kg単回投与後のFVIII活性の薬物動態パラメータ(12~65歳:発色合成基質法で測定)
| パラメータ | 日本人(N=10) | 全体(N=91) |
|---|---|---|
| IRa(IU/dL)/(IU/kg) | 2.07(11.2) | 1.97h(21.7) |
| Cmaxb(IU/dL) | 109(11.9) | 104h(19.3) |
| AUC0-infc(IU・hr/dL) | 2,060(15.9) | 1,910(34.0) |
| クリアランス(mL/hr/kg) | 2.49(17.1) | 3.00(37.7) |
| t1/2d(hr) | 16.4(29.0) | 14.2(26.6) |
| Vsse(mL/kg) | 55.7(20.5) | 56.7(23.5) |
| AUC0-lastf(IU・hr/dL) | 2,000(15.7) | 1,840(33.0) |
| MRTg(hr) | 23.0(25.4) | 20.4(26.4) |
a:投与量(IU/kg)当たりの投与後30分の上昇値、b:最高血中濃度、c:無限大まで外挿したFVIII活性-時間曲線下面積、d:半減期、e:定常状態分布容積、f:定量化可能な最終検体採取時刻までの活性-濃度曲線下面積、g:平均滞留時間、h:N=90
【対象及び方法】
血友病A[血液凝固第VIII因子(FVIII)活性<1%]患者を対象に、本剤50 IU/kgを単回投与後、ノンコンパートメントモデルを用いたPK解析を行った。
(2)反復投与後の薬物動態評価(日本人及び外国人データ)1)
エイフスチラ50 IU/kgの初回投与後薬物動態評価時及び反復投与後評価時において、各薬物動態パラメータは類似しており、初回投与時から反復投与時にかけての3~6ヵ月間で安定していた。
日本人(10例)で報告された平均値は全て全体で報告された値の範囲内であった。
エイフスチラ50 IU/kgの初回及び反復投与後のFVIII活性の薬物動態パラメータ(12~65歳:発色合成基質法で測定)
| パラメータ | 50 IU/kg初回(N=64) | 50 IU/kg反復(N=30) |
|---|---|---|
| IRa(IU/dL)/(IU/kg) | 1.85h(21.8) | 1.99i(17.7) |
| Cmaxb(IU/dL) | 99.9h(19.9) | 108i(17.2) |
| AUC0-infc(IU・hr/dL) | 1,830(34.9) | 1,880(34.5) |
| クリアランス(mL/hr/kg) | 3.15(38.2) | 3.05(36.0) |
| t1/2d(hr) | 14.1(27.1) | 12.9(29.4) |
| Vsse(mL/kg) | 59.5(23.9) | 53.1(16.4) |
| AUC0-lastf(IU・hr/dL) | 1,780(34.5) | 1,830(33.7) |
| MRTg(hr) | 20.3(26.4) | 18.9(28.5) |
a:投与量(IU/kg)当たりの投与後30分の上昇値、b:最高血中濃度、c:無限大まで外挿したFVIII活性-時間曲線下面積、d:半減期、e:定常状態分布容積、f:定量化可能な最終検体採取時刻までの活性-濃度曲線下面積、g:平均滞留時間、h:N=63、i:N=29
【対象及び方法】
血友病A[血液凝固第VIII因子(FVIII)活性<1%]患者64例を対象に、エイフスチラ50 IU/kgを単回投与後、初回PK評価を実施した。3~6ヵ月後、初回PK解析対象患者の部分集団30例を対象に、エイフスチラ50 IU/kgを単回投与し、再度PK評価を行った。初回及び反復PK解析はともにノンコンパートメントモデルを用いて行った。
3)小児患者の薬物動態評価(外国人データ)2)
エイフスチラ50 IU/kg単回投与後の薬物動態パラメータは、以下の通りであった。
エイフスチラ50 IU/kg単回投与後のFVIII活性の薬物動態パラメータ(12歳未満:発色合成基質法で測定)
| パラメータ | 6歳未満(N=20) | 6~12歳未満(N=19) |
|---|---|---|
| IRa(IU/dL)/(IU/kg) | 1.60(21.1) | 1.66(19.7) |
| Cmaxb(IU/dL) | 80.2(20.6) | 83.5(19.5) |
| AUC0-infc(IU・hr/dL) | 1,080(31.0) | 1,170(26.3) |
| クリアランス(mL/hr/kg) | 5.07(29.6) | 4.63(29.5) |
| t1/2d(hr) | 10.4(28.7) | 10.2(19.4) |
| Vsse(mL/kg) | 71.0(11.8) | 67.1(22.3) |
| AUC0-lastf(IU・hr/dL) | 1,010(28.4) | 1,090(26.4) |
| MRTg(hr) | 12.4(25.0) | 12.3(16.8) |
a:投与量(IU/kg)当たりの投与後60分の上昇値、b:最高血中濃度、c:無限大まで外挿したFVIII活性-時間曲線下面積、d:半減期、e:定常状態分布容積、f:定量化可能な最終検体採取時刻までの活性-濃度曲線下面積、g:平均滞留時間
【対象及び方法】
12歳未満の血友病A[血液凝固第VIII因子(FVIII)活性<1%]患者を対象に、本剤50 IU/kg単回投与後、ノンコンパートメントモデルを用いた年齢別(6歳未満20例、6~12歳未満19例)PK解析を行った。
2.分布3)
FVIIIは限定的な組織内分布を示し、大部分は血漿中に分布しており、同様なことが、ロノクトコグ アルファでも考えられる。また、ロノクトコグ アルファをカニクイザルに単回投与したときの分布容積は小さかった(VSS:26.6-40.3mL/kg)。
3.代謝
FVIIIは作用発現組織である血漿から消失する間に不活性になる4-6)ことから、代謝に関する試験は実施していない。
4.排泄
FVIIIは作用発現組織である血漿から消失する間に不活性になる4-6)ことから、排泄に関する試験は実施していない。
5.測定法による血液凝固第VIII因子活性値への影響7)
エイフスチラ投与後の血漿中血液凝固第VIII因子活性について、測定法(凝固一段法又は発色合成基質法)による測定値の相違を検討したところ、凝固一段法による測定値は発色合成基質法による測定値と比べて約45%低い値であった。
1)社内資料:日本人を含む国際多施設共同非盲検第I/III相試験(1001試験)(承認時評価資料)
2)社内資料:小児血友病A患者を対象とした海外多施設共同非盲検第III相試験(3002試験)(承認時評価資料)
3)社内資料:カニクイザルにおける単回静脈内投与薬物動態試験(承認時評価資料)
4)Oldenburg J et al.: Haemophilia. 2004; 10(Suppl 4): 133-139
5)Ananyeva NM et al.: Trends Cardiovasc Med. 2001; 11(6): 251-257
6)Lenting PJ et al.: Haemophilia. 2010; 16(Suppl 5): 194-199
7)社内資料:生物薬剤学試験及び関連する分析法(承認時評価資料)
8. 重要な基本的注意(一部抜粋)
8.3 十分な血液凝固第Ⅷ因子活性に到達・維持していることを確認するため、必要に応じ、血漿中血液凝固第Ⅷ因子活性をモニタリングすること。なお、本剤の活性(力価)は発色合成基質法により決定されているため、凝固一段法により本剤投与後の血漿中血液凝固第Ⅷ因子活性を測定した場合、測定結果が見かけ上低値を示すことが確認されている。本剤による治療中に血漿中血液凝固第Ⅷ因子活性を凝固一段法によりモニタリングする場合は、得られた血液凝固第Ⅷ因子レベルに換算係数2を乗じた値を用いること。[電子添文の7.1、16.8.1 参照]
JPN-AFS-1855
2024年6月改訂
薬効薬理 臨床薬理試験
1. 血漿中FVIII活性の推移(国際共同試験及び海外データ)1、2)
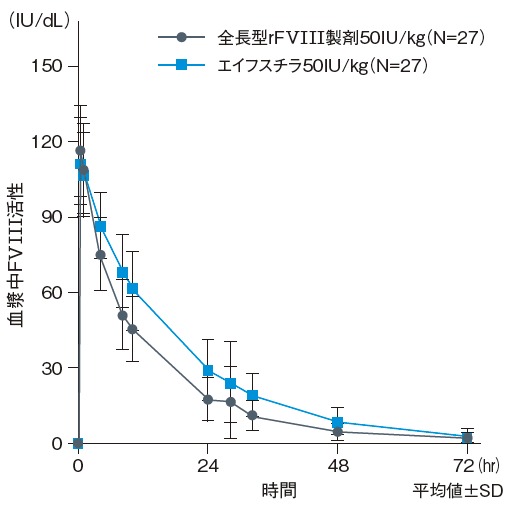
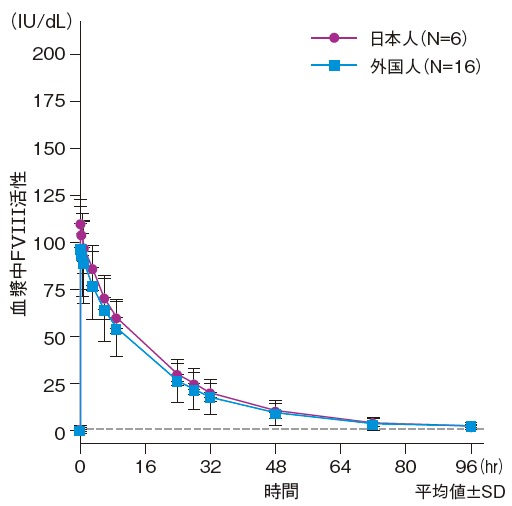
国際共同第I/III相試験1)のPK解析において、発色合成基質法により測定したエイフスチラ50 IU/kg及び全長型rFVIII製剤50 IU/kg単回投与後の最大FVIII活性の平均値は、エイフスチラでは113 IU/dL(中央値:109 IU/dL)、全長型rFVIII製剤では118 IU/dL(中央値:116 IU/dL)であった。その後、両製剤ともに血漿中FVIII活性は指数関数的に低下した。最終検体採取時点(投与後72時間)でのFVIII活性の平均値は、エイフスチラで4.19 IU/dL(中央値:3.86 IU/dL)、全長型rFVIII製剤で3.41 IU/dL(中央値:2.78 IU/dL)であった。なお、日本人及び外国人のFVⅢ活性プロファイルは以下の通りであった。
また、小児患者を対象とした海外第III相試験2)のPK解析において、色合成基質法により測定したエイフスチラ50 IU/kgIU/kg単回投与後の最大FⅤⅢ活性の平均値は、82.3 IU/dL(中央値:81.3 IU/dL)であった。最終検体採取時点(投与後48時間)でのFVIII活性の平均値は3.61 IU/dLであり、39例中32例がFVIII活性1%を超えていた。
単回投与後の血漿中FVIII活性の推移
エイフスチラ50 IU/kg単回投与後の血漿中FVIII活性の推移(日本人及び外国人)
【対象及び方法】
18~65歳の血友病A[血液凝固第VIII因子(FVIII)活性<1%]患者27例を対象に、全長型rFVIII製剤50 IU/kgを単回投与した際のFVIII活性を測定した後、4日間の休薬期間を経て、エイフスチラ50 IU/kgを単回静脈投与し、再びFVIII活性を測定した。
また、日本人及び外国人の比較では、12~65歳の患者(日本人6例、外国人16例)を対象に、エイフスチラ50 IU/kgを単回投与後、FVIII活性を測定した。FVIII活性はいずれも発色合成基質法により測定した。
2. 母集団PK解析を用いたFVIII活性シミュレーション3、4)
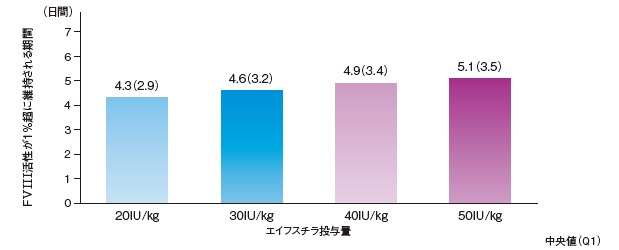
母集団PKモデルを用いて、エイフスチラ20~50 IU/kg単回投与後のFVIII活性が1%超に維持される期間のシミュレーションを行ったところ、エイフスチラ50 IU/kg単回投与後、約5.1日(中央値)でFVIII活性が約1 IU/dL(正常値の約1%)まで低下すると予測された。
また、エイフスチラ20~50 IU/kg反復投与後のFVIII活性についてシミュレーションを行ったところ、50 IU/kg週3回投与(Day 0、2及び4.5投与)において、12歳以上の患者群でのFVIII活性トラフ値(中央値)は、Day2、4.5、7で7.5、4.5、4.4 IU/dLと予測された。さらに、50 IU/kg週3回投与(Day 0、2及び4.5投与)で、Day 7におけるFVIII活性が1%を超える患者割合は93.2%と予測された。
エイフスチラ20~50 IU/kg単回投与後のFVIII活性が1%超に維持される期間
エイフスチラ20~50 IU/kg反復投与後のFVIII活性トラフ値が1%超に維持される患者の割合
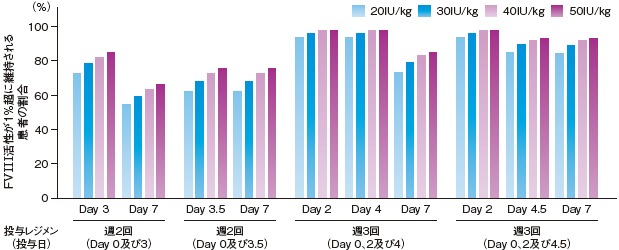
【対象及び方法】
エイフスチラの承認時臨床試験(2試験)から得られたPK総合データについて、0~65歳の患者計130例から得られた1,460測定点(日本人10例の208測定点)でのFVIII活性データを用いた2-コンパートメント母集団PKモデルにより、エイフスチラ20~50 IU/kg投与後の総FVIII活性のシミュレーションを行った。発色合成基質法により得られたFVIII活性データを基にしたシミュレーション結果を示した。
1)社内資料:日本人を含む国際多施設共同非盲検第I/III相試験(1001試験)(承認時評価資料)
2)社内資料:小児血友病A患者を対象とした海外多施設共同非盲検第III相試験(3002試験)(承認時評価資料)
3)社内資料:母集団PK解析(承認時評価資料)
4)Zhang Y et al.: J Thromb Haemost. 2017; 15(6): 1106-1114
JPN-AFS-1855
2024年6月改訂
薬効薬理 非臨床試験
(1)トロンビン生成に及ぼす影響(in vitro)11、12)
発色合成基質法で測定したFⅤⅢ活性に基づき、雌CDラット血漿にロノクトコグ アルファを1~30 IU/mLの濃度で添加し、トロンビン生成量を評価したところ、濃度10 IU/mLまで濃度依存的かつ統計学的に有意にトロンビンピーク値が上昇した(下表)。また、発色合成基質法により測定した乏血小板血漿に対するFⅤⅢ活性に基づき、カニクイザルの血漿にロノクトコグ アルファを1~30 IU/mLの濃度で添加し、トロンビン生成量を評価したところ、最高濃度30 IU/mLまで濃度依存的かつ統計学的に有意にトロンビンピーク値及び内因性トロンビン活性が増加し(下表)、ピーク値到達時間が短縮した。
ロノクトコグ アルファ添加後のラット血漿中のトロンビンピーク値の群間差
| 群 | 比較対象群 | 平均値差の推定値 (トロンビン、nmol) | 標準誤差 (トロンビン、nmol) | 両側p値* |
|---|---|---|---|---|
| 1 IU/mL | 陰性対照群 | 12.9 | 3.87 | 0.0205 |
| 3 IU/mL | 陰性対照群 | 24.6 | 3.24 | 0.0006 |
| 10 IU/mL | 陰性対照群 | 39.7 | 2.81 | < 0.0001 |
| 30 IU/mL | 陰性対照群 | 44.8 | 8.12 | 0.0027 |
| 3 IU/mL | 1 IU/mL | 9.8 | 2.23 | 0.0071 |
| 10 IU/mL | 1 IU/mL | 24.4 | 3.78 | 0.0013 |
| 30 IU/mL | 1 IU/mL | 30.7 | 7.04 | 0.0072 |
| 10 IU/mL | 3 IU/mL | 14.4 | 4.32 | 0.0207 |
| 30 IU/mL | 3 IU/mL | 21.2 | 4.58 | 0.0057 |
| 30 IU/mL | 10 IU/mL | 4.8 | 5.55 | 0.43 |
ロノクトコグ アルファ添加後のカニクイザル血漿中のトロンビンピーク値の群間差
| 群 | 比較対象群 | 平均値差の推定値 (トロンビン、nmol) | 標準誤差 (トロンビン、nmol) | 両側p値* |
|---|---|---|---|---|
| 1 IU/mL | 陰性対照群 | 11.0 | 11.34 | 0.36 |
| 3 IU/mL | 陰性対照群 | 20.7 | 7.90 | 0.0343 |
| 10 IU/mL | 陰性対照群 | 64.7 | 10.39 | 0.0004 |
| 30 IU/mL | 陰性対照群 | 95.8 | 7.54 | < 0.0001 |
| 3 IU/mL | 1 IU/mL | 9.7 | 6.45 | 0.18 |
| 10 IU/mL | 1 IU/mL | 53.7 | 5.29 | < 0.0001 |
| 30 IU/mL | 1 IU/mL | 84.8 | 6.59 | < 0.0001 |
| 10 IU/mL | 3 IU/mL | 44.0 | 4.96 | < 0.0001 |
| 30 IU/mL | 3 IU/mL | 75.1 | 3.97 | < 0.0001 |
| 30 IU/mL | 10 IU/mL | 31.1 | 5.37 | 0.0007 |
【実験方法】
発色合成基質法で測定したFVⅢ活性に基づき、雄CDラット血漿およびカニクイザル血漿にロノクトコグ アルファを1~30 IU/mLの濃度で添加した。PPP試薬をトリガーとして凝固を誘発し、自動較正トロンボグラム(CAT)分析によりトロンビンピーク値を解析した。
2. トロンボエラストグラフィ及びトロンビン生成による血液凝固活性の検討[血友病Aマウス(FVIII欠損マウス)]3)
FVIII欠損マウス(雄5匹、雌5~6匹)にロノクトコグ アルファ及びルリオクトコグ アルファを発色合成基質法で測定したFVIII活性に基づき20 IU/kgの用量で静脈内投与し、トロンボエラストグラフィ(TEG)及びトロンビン生成パラメータの解析を行った。溶媒対照群と比較して、両群ともにTEGパラメーターが改善し、止血作用の回復を示唆する効果が認められた。
3. 凝固パラメータ(aPTT)に対する作用(FVIII欠損マウス)4)
FVIII欠損マウスを用いて、ロノクトコグ アルファ又はルリオクトコグ アルファによる活性化部分トロンボプラスチン時間(aPTT)への作用を検討したところ、対照群と比較して、両群ともにaPTTが短縮し、止血作用の回復を示唆する効果が認められた。
ロノクトコグ アルファ又はルリオクトコグ アルファ単回静脈内投与後のaPTT値に対する作用
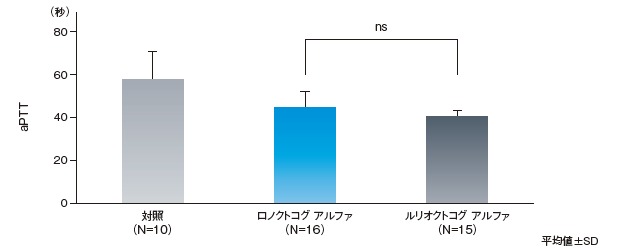
【実験方法】
FVIII欠損マウス(雄5~8匹、雌5~10匹)にロノクトコグ アルファは発色合成基質法で測定したFVIII活性に基づく用量、ルリオクトコグ アルファは表示力価に基づく用量でそれぞれ20 IU/kg単回静脈内投与した。その15分後に採血し、止血効果を評価するため凝固パラメータ(aPTT)を解析した。
4. 止血作用(FVIII欠損マウス)5、6)
FVIII欠損マウスのテイルクリップ・モデルを用いてrFVIII製剤の止血効果を検討したところ、ロノクトコグ アルファ群では、有意かつ用量依存的な総失血量の減少(p<0.0001、共分散分析、共変量:性別、対数用量)、止血までの時間及びaPTTの短縮(p<0.0001、線形ロジスティック回帰分析、共変量:性別、用量段階)が認められた。
ロノクトコグ アルファ又は市販の臨床用rFVIII各製剤の用量依存的な止血効果a
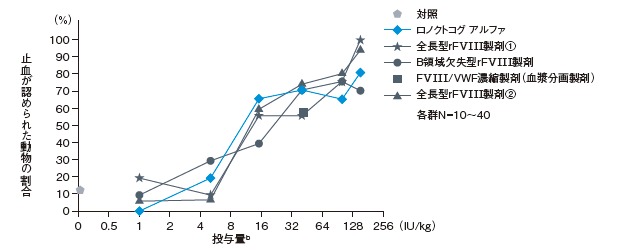
b:発色合成基質法で測定したFVIII活性に基づく投与量
【実験方法】
FVIII欠損マウス(雄5~15匹、雌5~15匹/群)にロノクトコグ アルファ又は市販の臨床用rFVIII各製剤[全長型製剤2製剤、B領域欠失型製剤、FVIII/VWF濃縮製剤(血漿分画製剤)]※を発色合成基質法で測定したFVIII活性に基づき1~150 IU/kgの用量(FVIII/VWF濃縮製剤は41 IU/kg)で静脈内投与した。テイルクリップ・モデルを用いて止血効果を測定した。
※本試験で使用した臨床用rFVIII製剤は全長型rFVIII製剤②を除き、国内未承認
1)社内資料:ラット血漿中トロンビン生成を指標とした薬力学的効果の評価(承認時評価資料)
2)社内資料:サル血漿中トロンビン生成を指標とした薬力学的効果の評価(承認時評価資料)
3)社内資料:第VIII因子ノックアウトマウスにおけるトロンボエラストグラフィ及びトロンビン生成(承認時評価資料)
4)社内資料:第VIII因子ノックアウトマウスにおけるaPTTに対する効果(承認時評価資料)
5)社内資料:第VIII因子ノックアウトマウスにおける止血効果(4市販製剤との比較)(承認時評価資料)
6)社内資料:第VIII因子ノックアウトマウスにおける止血効果(5市販製剤との比較)(承認時評価資料)
JPN-AFS-1855
2024年6月改訂
調製方法安全性薬理試験および毒性試験
1.調製方法
※作業は全て無菌的に行ってください。
(1)溶解準備
- エイフスチラを冷蔵庫から取り出した場合は、室温に戻しておく。25℃を超えて保存した場合、25℃以下で3ヵ月を超えて保存した場合は使用しない。
- 作業場所(水平な台)を清潔にし、手を石けんでしっかり洗う。
- 使用前にバイアルに異常(変色、濁り、容器の破損など)がないか確認する。
- 薬剤バイアルと溶解液バイアルのキャップを外し、ゴム栓をアルコール綿で拭く。
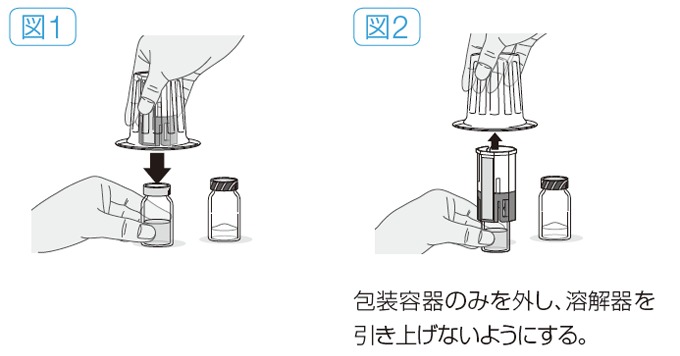
(2)バイアルのセット
- 専用溶解器(スリートック通気孔タイプ)の包装容器のふたをはがす。
- 溶解器を包装容器ごと持ち、青色側の穿刺部を溶解液バイアルのゴム栓にまっすぐ下向きに刺し込む(図1)。
- 包装容器を溶解器から慎重に外す(図2)。
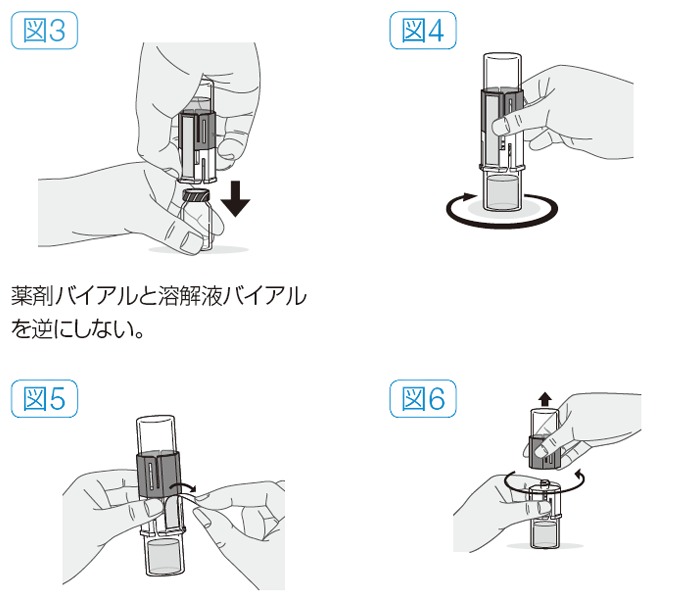
(3)薬剤の溶解
- 溶解液バイアルを逆さまにし、透明側の穿刺部を下に置いた薬剤バイアルのゴム栓に、まっすぐ下向きに一気に刺し込み、溶解液を薬剤バイアル中に移行させる(図3)。
- そのまま台の上で、薬剤バイアルをゆるやかに、小さく円を描くように回し(激しく回さない)、完全に薬剤を溶かす(図4)。
- 溶解器側面のシールをはがす(図5)。
- 溶解器の青色側の部分を反時計回りに回して2つに分ける(図6)。
(4)薬液を注射器に移す
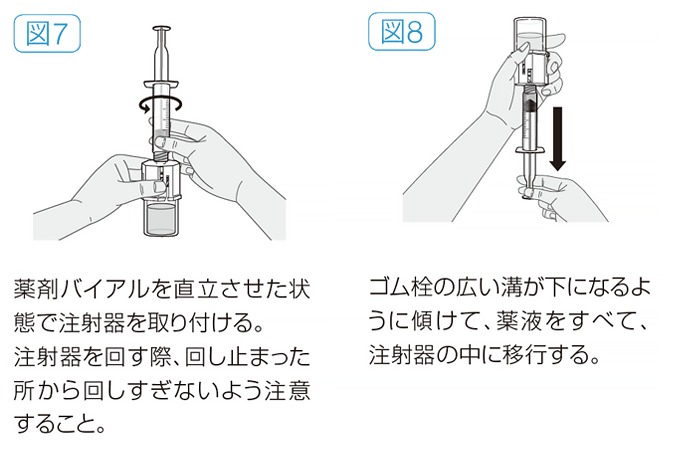
- 空の滅菌済注射器に空気が入っていないことを確認し、溶解器のルアーロックの奥まで注射器を時計回りに回しながら取り付ける(図7)。
- 薬剤バイアルを上にして、プランジャーロッドをゆっくりと引き、薬液を注射器の中に移行する(図8)。
- すべての薬液を注射器の中に移行したら、注射器を下にしたまま、溶解器を反時計回りに回して注射器から外す。
(5)注射器に翼状針を取り付ける
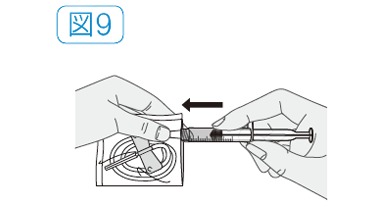
- 翼状針の接続部分が袋の切り口近くにくるように、翼状針の袋を開ける。
- 薬液を移行した注射器を、翼状針の接続部分に差し込み、取り付ける(図9)。
- 袋を外し、注射器のプランジャーロッドを少し押して、薬液を針先まで満たす。
2.一般薬理試験及び毒性試験
1.一般薬理試験(ラット、イヌ、サル)1-5)
| 試験項目 | 動物種/ 系統 | 性別/ 匹数/ 各投与群 | 投与期間/ 投与経路 | 投与量 (IU/kg) | 試験結果 | |
|---|---|---|---|---|---|---|
| 中枢神経系 への影響 | 一般状態、 Irwinの変法による 神経行動学的観察評価、 病理組織学的観察、 肉眼的観察 | ラット/SD | 雄10、雌10 | 4週間反復a/ 静脈内 | 0、50、 250、 1,250 | 本剤による影響なし 無毒性量:1,250 IU/kg |
| 心血管系 への影響 | 血圧、心拍数、心電図、 左室圧パラメータ及び その派生パラメータ、 心拍出量、1回拍出量、 末梢抵抗 | イヌ/ ビーグル犬 | 雄4 | 45分間隔で 30分間の投与 が3回/静脈内 | 0又は 1,550b | 3回目の投与後、心血管系 パラメータ(動脈血圧、左 室圧パラメータ及びその派 生パラメータ、心拍出量、 1回拍出量)の低下 |
| 一般状態、血圧、心電図、 心拍数、左室圧パラメータ 及びその派生パラメータ、 dP/dt(テレメーター装着) | イヌ/ ビーグル犬 | 雄1 | 45分間隔で 30分間の投与 が3回/静脈内 | 0又は 1,550b | 本剤による影響なし | |
| 肉眼的観察、 病理組織学的観察、心電図、 心拍数、血圧、一般状態、 検眼鏡検査、血液学的検査、 血液化学検査、尿検査 | サル/ カニクイザル | 雄4、雌4 | 4週間反復/ 静脈内 | 0、50、 150、 500 | 本剤の投与による明らかな 影響は認められなかった 5日目の投与2時間後に、 500 IU/kg/日群の雄で平均 脈拍数の軽微な減少あり 無毒性量:500 IU/kg | |
| 一般状態、心電図、心拍数、 血圧(テレメーター装着) | サル/ カニクイザル | 雄2、雌2 | 45分間隔で 30分間の投与 が3回/静脈内 | 0又は 1,550b | 本剤による影響なし | |
| 呼吸器系 への影響 | 呼吸数、1回換気量、 分時換気量 | イヌ/ ビーグル犬 | 雄4 | 45分間隔で 30分間の投与 が3回/静脈内 | 0又は 1,550b | 本剤による影響なし |
b:50(初回)、250(2回目)及び1,250 IU/kg(3回目)による漸増投与の累積投与量
3.毒性試験
(1)単回投与毒性試験(ラット、サル)6、7)
| 動物種/ 系統 | 性別/ 匹数 | 投与経路 | 投与量 (IU/kg) | 無毒性量/ 概略致死量 | 試験結果 |
|---|---|---|---|---|---|
| ラット/SD | 雄5、雌5 | 静脈内 | 0、50、250、1,500 | 1,500 IU/kg />1,500 IU/kg | 毒性所見なし |
| サル/ カニクイザル | 雄3、雌3 | 静脈内 | 0、50、250、1,500 | 1,500 IU/kg />1,500 IU/kg | 毒性所見なし |
(2)反復投与毒性試験(ラット、サル)8、9)
| 動物種/ 系統 | 性別/匹数 | 投与期間/ 投与経路 | 投与量 (IU/kg) | 無毒性量 | 試験結果 |
|---|---|---|---|---|---|
| ラット/SD | 0及び1,250 IU/kg:雄20、雌20 50及び250 IU/kg:雄15、雌15 | 4週間a/ 静脈内 | 0、50、250、1,250 | 1,250 IU/kg | 毒性所見なし |
| サル/ カニクイザル | 雄4、雌4 | 4週間a/ 静脈内 | 0、50、150、500 | 500 IU/kg | 毒性所見なし |
3)局所刺激性試験(ウサギ)10)
| 動物種/系統/性別/匹数(各群) | 投与量(投与方法) | 試験結果 |
|---|---|---|
| ウサギ/NZW/雄3 | 1.0mL/匹(静脈内) 1.0mL/匹(動脈内) 0.2mL/匹(静脈周囲) | 投与局所の所見及び全身症状なし |
4)血栓形成性試験a(ウサギ)11)
| 動物種/系統/性別/匹数(各群) | 投与量(投与方法) | 試験結果 |
|---|---|---|
| ウサギ/NZW/雄3、雌3b | 150、300、500、1,000 IU/kg (静脈内) | 1,000 IU/kgで軽微な血栓形成 無毒性量:500 IU/kg |
b:対照群として雌雄各6匹に生理食塩液を投与
1)社内資料:ラットにおける4週間静脈内投与毒性試験(承認時評価資料)
2)社内資料:カニクイザルにおける4週間静脈内投与毒性試験(承認時評価資料)
3)社内資料:麻酔ビーグル犬における血行動態及び呼吸機能への影響(承認時評価資料)
4)社内資料:覚醒ビーグル犬における心血管系テレメトリー試験(承認時評価資料)
5)社内資料:覚醒カニクイザルにおける心血管系テレメトリー試験(承認時評価資料)
6)社内資料:ラットにおける単回投与毒性試験(承認時評価資料)
7)社内資料:カニクイザルにおける単回投与毒性試験(承認時評価資料)
8)社内資料:ラットにおける4週間静脈内投与毒性試験(承認時評価資料)
9)社内資料:カニクイザルにおける4週間静脈内投与毒性試験(承認時評価資料)
10)社内資料:ウサギにおける局所刺激性試験(承認時評価資料)
11)社内資料:ウサギにおける血栓形成性試験(承認時評価資料)
JPN-AFS-1855
2024年6月改訂
製剤学的事項
1.製剤の安定性
エイフスチラ静注用の各種条件下における安定性
| 試験 | 保存条件 | 実施期間 | 試験結果 |
|---|---|---|---|
| 長期保存試験a | 5±3℃/遮光保存 | 36ヵ月 | 全ての試験項目で規格内 |
| 加速試験 | 30±2℃/75±5%RH | 12ヵ月 | FVIII活性の低下、目的物質由来不純物の 増加及び目的物質の減少が認められた。 |
| 苛酷試験 (温度) | 40±2℃/75±5%RH | 6ヵ月 | |
| 苛酷試験b (光) | 25℃/60%RH/総照度: 120万lux・h以上、 総近紫外放射エネルギー: 200W・h/m²以上 | 1週間 | FVIII活性の低下 |
| 溶解後安定性試験 | 20~25℃ | 溶解後4時間 | 全ての試験項目で規格内 |
b:1ロットの試験実施
<測定項目>
長期保存試験:性状、確認試験、浸透圧、pH、純度、水分、エンドトキシン、不溶性異物、不溶性微粒子、無菌、溶解時間、比活性、定量
加速試験:性状、確認試験、pH、純度、水分、エンドトキシン、不溶性異物、無菌、溶解時間、比活性、定量
苛酷試験(温度):性状、確認試験、pH、純度、水分、エンドトキシン、不溶性異物、溶解時間、比活性、定量
苛酷試験(光):性状、pH、純度、不溶性異物、不溶性微粒子、溶解時間、比活性、定量
溶解後安定性試験:性状、確認試験、pH、純度、比活性、定量
2.製造工程
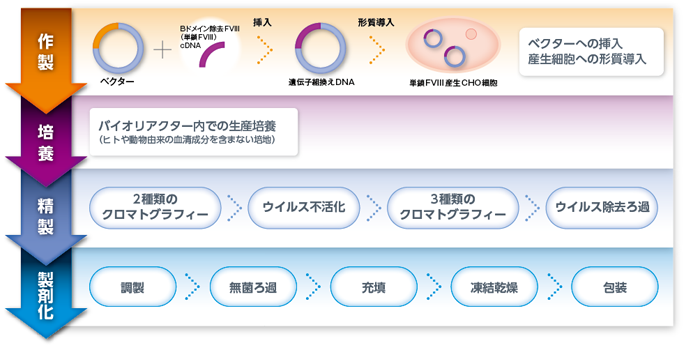
FVIII:血液凝固第VIII因子 CHO:チャイニーズハムスター卵巣
JPN-AFS-1855
2024年6月改訂
取扱い上の注意
| 規制区分 | 生物由来製品、処方箋医薬品(注意-医師等の処方箋により使用すること) |
|---|---|
| 貯法 | 2~8℃で保存 |
| 有効期間 | 36箇月 |
| 取り扱い上の注意 | 外箱開封後は遮光して保存すること。 |
JPN-AFS-1855
2024年6月改訂
災害時の製品保管について
停電から復旧後は、冷蔵庫には戻さず、室温(25℃以下)にて使用期限を超えない範囲で3ヵ月以内にご使用ください。
エイフスチラ静注用の各種条件下における安定性
| 試験 | 保存条件 | 実施期間 | 試験結果 |
|---|---|---|---|
| 長期保存試験a | 5±3℃/遮光保存 | 36ヵ月 | 全ての試験項目で規格内 |
| 加速試験 | 30±2℃/75±5%RH | 12ヵ月 | FVIII活性の低下、目的物質由来不純物の増加 及び目的物質の減少が認められた。 |
| 苛酷試験 (温度) | 40±2℃/75±5%RH | 6ヵ月 | |
| 苛酷試験b (光) | 25℃/60%RH/総照度: 120万lux・h以上、 総近紫外放射エネルギー: 200W・h/m²以上 | 1週間 | FVIII活性の低下 |
| 溶解後安定性試験 | 20~25℃ | 溶解後4時間 | 全ての試験項目で規格内 |
b:1ロットの試験実施
<測定項目>
長期保存試験:性状、確認試験、浸透圧、pH、純度、水分、エンドトキシン、不溶性異物、不溶性微粒子、無菌、溶解時間、比活性、定量
加速試験:性状、確認試験、pH、純度、水分、エンドトキシン、不溶性異物、無菌、溶解時間、比活性、定量
苛酷試験(温度):性状、確認試験、pH、純度、水分、エンドトキシン、不溶性異物、溶解時間、比活性、定量
苛酷試験(光):性状、pH、純度、不溶性異物、不溶性微粒子、溶解時間、比活性、定量
溶解後安定性試験:性状、確認試験、pH、純度、比活性、定量
2024年6月改訂