臨床成績 12歳未満の小児に対する海外多施設共同非盲検第III相試験(3002試験、海外データ)1)
1. 試験概要
試験デザイン
海外、多施設共同(37施設)、プロスペクティブ、非盲検、第III相試験
目的
重症の小児血友病A患者を対象に、以下の項目について評価する。
- 出血エピソードの抑制及び治療の有効性
- 定期補充療法及びオンデマンド療法の有効性
- エイフスチラの投与量
- エイフスチラの薬物動態(PK)プロファイル
- FVIIIインヒビターの発現
- エイフスチラの安全性プロファイル
対象
12歳未満の重症(FVIII活性<1%)血友病A患者 84例(6歳未満35例、6~12歳未満49例)
- FVIIIインヒビター発現の既往が確認されていない患者
- FVIII製剤の投与日数が50EDを超える12歳未満の男性患者
方法
スクリーニング期間28日間、PK評価期間2日間、治療期間約6ヵ月(各患者50ED以上)から構成された。
| 定期補充療法 | エイフスチラを1日おきもしくは週2~3回15~50 IU/kg、又は過去のFVIII用量及びPKデータに基づき治験責任医師が決定した用量及び頻度で投与した。 |
|---|---|
| オンデマンド療法 | エイフスチラを治験責任医師が判断した用量で投与した。 |

主要評価項目
有効性
4点スケール(「著効」、「有効」、「やや有効」、「無効/反応なし」)に基づいた治験責任医師による出血エピソードの治療成功率※
※ 総合的臨床評価が「著効」又は「有効」である場合
副次評価項目
薬物動態(PK)
エイフスチラ50 IU/kg単回投与後のPK[投与後60分の上昇値(IR)、半減期(t1/2)など]
有効性
- 定期補充療法及びオンデマンド療法での年間自然出血回数(AsBR)、年間総出血回数(総ABR)及びエイフスチラの投与量
- 止血が得られるまでの投与回数及びエイフスチラの投与量
安全性
ナイメゲン変法を用いたベセスダ測定に基づくインヒビターの発現(0.6BU/mL以上)、有害事象、臨床検査値、身体検査所見、バイタルサイン
解析手法
出血エピソードの止血効果:治療が成功(止血効果の評価が「著効」又は「有効」)であった出血エピソードの割合である治療成功率及び95%CIを算出した。主要解析では、治験責任医師の評価のない治療された出血エピソードは治療不良とみなした。患者内相関を考慮し、二項分布とリジットリンク関数を仮定した反復測定に対する一般化線形モデル(切片項のみを含む)を用いた。止血効果に関する治験責任医師の総合的臨床評価について、有効性解析集団での年齢、BMI、人種及び地域別サブグループ解析も実施した。
年間出血回数:ABRは、365.25×(治療を要した出血エピソード回数/有効性評価日数)と定義した。総出血、自然出血、外傷性出血、及び関節出血のABRを算出し、記述統計量を用いて要約した。また、Poisson回帰モデルに基づき、出血エピソード(総出血、自然出血、外傷性出血、及び関節出血)の年間発現回数及び95%CIを算出した。ABRは、治療法(オンデマンド療法又は定期補充療法)別に示し、年齢、BMI、人種及び地域別サブグループ解析も実施した。
有効性解析集団
試験期間中にエイフスチラの投与を1回以上受け、登録時にインヒビター陰性であった患者 83例
安全性解析集団
試験期間中にエイフスチラの投与を1回以上受けた全ての患者 84例
ED:投与日(Exposure day)
評価基準[出血エピソードに対する止血効果]
小出血
| 著効 | 本剤を最初に投与してから約8時間以内に明確な疼痛緩和があった、及び/又は出血の徴候が改善した(腫脹、圧痛の改善及び/又は筋骨格系出血の場合には可動域の増加)。 |
|---|---|
| 有効 | 本剤を最初に投与してから約8時間後の時点で明確な疼痛緩和があった、及び/又は出血の徴候が改善したが、完全な消失には2回の投与を要する。 |
| やや有効 | 本剤を最初に投与してから約8時間以内にわずかに有益な効果が見られた、及び/又は推定されたが、完全な消失には2回を超える投与を要する。 |
| 無効/反応なし | 本剤を最初に投与してから全く改善が見られない、又は状態(出血の徴候)が悪化し、完全な消失には他の第VIII因子製剤、クリオプレシピテート又は血漿を用いた追加の止血治療を要する。 |
大外傷又は生命を脅かす出血
| 著効 | 止血の程度について、凝固因子欠乏を有しない患者と比べて臨床的に有意な差がなく(同程度の止血が得られる)、推定失血量が同程度の外傷又はその他要因から予想される推定失血量と比べて20%以上増加しない。 |
|---|---|
| 有効 | 止血の程度について、凝固因子欠乏を有しない患者と比べて、正常又は軽度な異常がある(わずかな滲出、出血量が若干多く止血までの時間が長い)、又は推定失血量が同程度の外傷又はその他要因から予想される推定失血量と比べて20%~30%増加している。 |
| やや有効 | 止血の程度について、凝固因子欠乏を有しない患者と比べて、正常又は軽度な異常がある(わずかな滲出、出血量が若干多く止血までの時間が長い)、又は推定失血量が同程度の外傷又はその他要因から予想される推定失血量と比べて20%~30%増加している。中等度の異常がある(コントロールが難しい中等度の出血)、推定失血量が「有効」と比べて多い。 |
| 無効/反応なし | 止血の程度について、凝固因子欠乏を有しない患者と比べて、重度の異常がある(コントロールが難しい重度の出血)、及び/又は同程度の外傷又はその他要因から予想される以上に、追加の他の第VIII因子製剤、クリオ製剤又は血漿による止血処置を必要とする。 |
2. 出血時の止血に対する有効性
1)止血効果の臨床評価(主要評価項目)
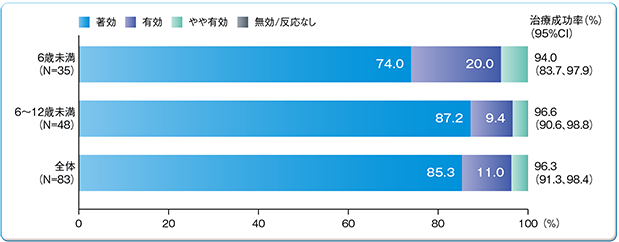
有効性解析集団(83例)において、治療を要した出血エピソード数は347件であった。治験責任医師による止血効果の総合的臨床評価は、大部分が「著効」(296/347件、85.3%)もしくは「有効」(38/347件、11.0%)であり、治療成功率※(95%CI)は96.3(91.3、98.4)%であった。年齢別の治療成功率は6歳未満で94.0(83.7、97.9)%、6歳以上12歳未満で96.6(90.6、96.8)%であった。
※「著効」又は「有効」を治療成功とした。95%CIは患者内相関を考慮した一般化線形モデルに基づく。
治験責任医師による出血に対する止血効果の総合的な臨床評価
2)止血が得られるまでの投与回数及び投与量(副次評価項目)
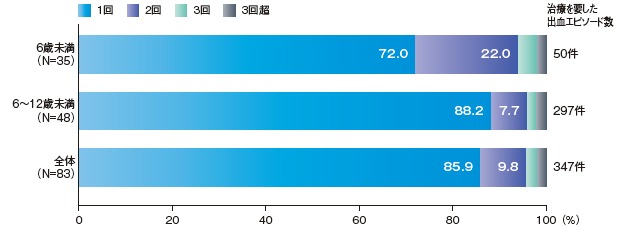
有効性解析集団(83例)において、治療を要した出血エピソード347件のうち332件(95.7%)はエイフスチラの1回又は2回投与で止血が得られた。
止血を得るのに要した1回当たりのエイフスチラ投与量及び総投与量[中央値(最小値、最大値)]は27.3(16、76)IU/kg及び27.6(16、282)IU/kgであった。
止血を得るのに要したエイフスチラの投与回数
3. 定期補充療法に対する有効性
1)定期補充療法における年間自然出血回数及び年間総出血回数(副次評価項目)
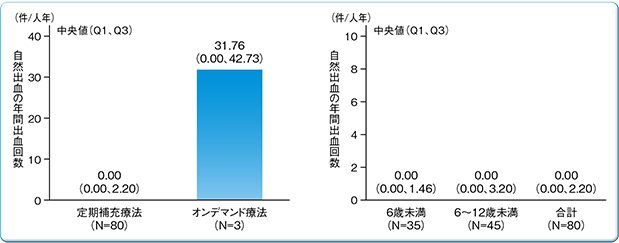
有効性解析集団(83例)において、定期補充療法による年間自然出血回数(AsBR)及び年間総出血回数(総ABR)の中央値[第1四分位値(Q1)、第3四分位値(Q3)]は、0.00(0.00、2.20)件/人年及び3.69(0.00、7.20)件/人年であった。年齢別のAsBR及び総ABRは以下の通りであった。
なお、定期補充療法に割り付けられた80例中67例(84%)が週2~3回投与であった。
定期補充療法における自然出血の年間出血回数(AsBR)
出血の種類別年間出血回数
| 全体の有効性解析集団 | 年齢別(定期補充療法群) | |||
|---|---|---|---|---|
| 年間出血回数 (件/人年) | 定期補充療法 (N=80) | オンデマンド療法 (N=3) | 6歳未満 (N=35) | 6~12歳未満 (N=45) |
| 自然出血(AsBR) 平均値(SD) 中央値 (Q1、Q3) | 1.70(2.972) 0.00 0.00、2.20 | 24.83(22.191) 31.76 0.00、42.73 | 0.81(1.499) 0.00 0.00、1.46 | 2.39(3.607) 0.00 0.00、3.20 |
| 総出血(総ABR) 平均値(SD) 中央値 (Q1、Q3) | 5.22(5.557) 3.69 0.00、7.20 | 66.77(27.702) 78.56 35.12、86.62 | 3.00(3.932) 2.12 0.00、4.54 | 6.94(6.047) 5.11 2.52、10.50 |
| 出血回数が0回であった患者数 | 21(26.3%) | 0 | 14(40.0%) | 7(15.6%) |
2)定期補充療法期間中のエイフスチラの投与量(副次評価項目)
定期補充療法を受けた患者80例のエイフスチラの月間及び年間投与量の中央値は、342 IU/kg及び4,109 IU/kgであった。また、週3回投与群(24例)及び週2回投与群(43例)での1回当たりのエイフスチラ投与量の中央値は、32.0 IU/kg及び35.0 IU/kgであった。
4. 安全性
1)インヒビター及び抗体(副次評価項目)
安全性解析集団においてインヒビター発現率(95%CI)は0(0、4.3)%※であった。また、CHO宿主細胞由来タンパク質に対する抗体の発現が報告された症例はなかった。
※試験登録前から低力価インヒビターが存在した1例を除く。
2)副作用(副次評価項目)
本剤の投与期間中、安全性解析集団84例中64例(76.2%)に183件の有害事象が認められた。副作用(本剤に関連する有害事象)は84例中1例(1.2%)に1件(過敏症)認められた。重篤な有害事象は84例中9例(10.7%)に11件[裂傷(1件)、医療機器閉塞(1件)、貧血(3件)、消化不良(1件)、菌血症(1件)、肺炎(1件)、全身性炎症反応症候群(1件)、手骨折(1件)、脾破裂(1件)]認められたが、いずれも本剤投与との因果関係はないと判断された。投与中止に至った症例及び死亡例はなかった。
1)社内資料:小児血友病A患者を対象とした海外多施設共同非盲検第III相試験(3002試験)(承認時評価資料)
6. 用法及び用量
本剤を添付の溶解液全量で溶解し、緩徐に静脈内に注射する。
通常、1回体重1kg当たり10~30国際単位を投与するが、患者の状態に応じて適宜増減する。
定期的に投与する場合、通常、体重1kg当たり20~50国際単位を週2回又は週3回投与する。
8. 重要な基本的注意(一部抜粋)
8.2 患者の血中に血液凝固第IX因子に対するインヒビターが発生するおそれがある。特に、血液凝固第Ⅷ因子製剤による補充療法開始後、投与回数が少ない時期(補充療法開始後の比較的早期)や短期間に集中して補充療法を受けた時期にインヒビターが発生しやすいことが知られている。本剤を投与しても予想した止血効果が得られない場合には、インヒビターの発生を疑い、回収率やインヒビターの検査を行うなど注意深く対応し、適切な処置を行うこと。
9. 特定の背景を有する患者に関する注意(一部抜粋)
9.1 合併症・既往歴等のある患者
9.1.1 本剤の有効成分及び添加剤、又はハムスター由来蛋白質に対し過敏症の既往歴のある患者
9.1.2 他の血液凝固第Ⅷ因子製剤に対し過敏症の既往歴のある患者
9.7 小児等
投与量及び投与頻度の調整について適宜検討すること。0~12歳未満の小児では、体重当たりのクリアランスが高値であり、通常よりも高い投与量及び頻回の投与が必要となる可能性がある。[電子添文の16.1.2参照]
2024年6月改訂