2.禁忌(次の患者には投与しないこと)
本剤の成分に対し過敏症の既往歴のある患者
本試験には一部承認用量よりも高用量(80 IU/kg)を投与した症例が含まれます。承認時評価資料であり、原著との不整合が生じることを防ぐために承認外用量を含む内容を記載しました。また、本試験には一部承認外の用量(40 IU/kg週2回皮下投与)を投与した症例が含まれますが、承認の範囲内の症例群のみに限定し、一部改変しました。
海外第III相臨床試験:COMPACT長期投与試験(海外データ)1,2)
1)社内資料:海外第III相試験(CSL830_3002)(CTD 2.7.6.4)(承認時評価資料)
2)Craig T, et al.:J Allergy Clin Immunol Pract.2019;7(6):1793-1802
利益相反:本試験はCSLベーリングより資金助成を受けた。著者にCSLベーリングよりコンサルト料、謝礼、
研究費を受領している者が含まれる。本論文の著者のうち5人はCSLベーリングの社員である。
試験概要
目的
遺伝性血管性浮腫(HAE)の長期予防を目的としてベリナート皮下注用を皮下投与したときの安全性を評価する。
対象
HAE 1型又は2型と確定診断された6歳以上のHAE患者126例
[COMPACT試験に参加した64例(継続例12例+中断例52例)及び非参加の未投与例62例]
| 継続例 | COMPACT試験を完了し、本試験へ直接(COMPACT試験の試験終了来院から本試験の治療期1の初回来院まで1週間以内に)参加した被験者 |
|---|---|
| 中断例 | COMPACT試験を完了したものの本試験へ遅れて(COMPACT試験の試験終了来院から本試験の初回来院[スクリーニング来院]まで1週間超経過後)参加した被験者 |
| 未投与例 | 先行試験であるCOMPACT試験に参加しなかった又はCOMPACT試験に参加したが試験期間中に本剤を投与されなかった被験者 |
試験デザイン
多施設共同、ランダム化、非盲検、並行群間比較、第IIIb相試験
方法
スクリーニング期、治療期1、治療期2、継続投与期及び追跡調査期の5パートで構成された。
- スクリーニング期(最長4週間)
中断例及び未投与例に対しては、スクリーニング来院を実施して適格性を確認した。継続例はスクリーニング来院を実施せず、代わりにCOMPACT試験のデータを適格性確認に使用した。適格被験者は、40 IU/kg又は60 IU/kgのいずれかを固定用量として24週間投与する治療期1へランダムに割り付けた。 - 治療期1(固定用量期:24週間)
HAE発作が頻回(4週間の評価期間中に12回以上)発現した被験者には、20 IU/kgずつ(最大80 IU/kgまで)漸増可能とした。
40 IU/kg投与群
ベリナート皮下注用40 IU/kgを週2回皮下投与。
60 IU/kg投与群
ベリナート皮下注用60 IU/kgを週2回皮下投与。 - 治療期2(用量調節期:28週間)
両群ともweek 25から個別に長期予防を最適化することが可能であり、8週間の評価期間中に3回以上HAE発作が発現した被験者には、20 IU/kgずつ(最大80 IU/kgまで)漸増可能とした。 - 継続投与期(最長88週間)
米国限定で、治療期2を完了した被験者は継続投与期へ任意に移行し、非盲検で治療を継続することができた。 - 追跡調査期(2週間)
同意/アセントが撤回されない限り、治療期2もしくは継続投与期(該当する場合)の最終来院、又は試験中止時来院から14(±3)日後に追跡調査来院を実施した。

評価項目
| 主要評価項目 | 以下の各項目の人年法による発現率(PTIR)[試験の早期中止に至った有害事象、血栓塞栓性事象、アナフィラキシー、入院を要したHAE発作、治験責任医師が重度と判定した注射部位反応、本剤との因果関係ありと判定された重篤な有害事象、抗C1-INH抗体] |
|---|---|
| 副次評価項目 | 有害事象、重篤な有害事象、HAE発作頻度が50%以上相対的に減少した被験者(レスポンダー)の割合、4週あたりの期間で調整したHAE発作頻度が1回未満であった被験者の割合 等 |
| 探索的評価項目 | 期間で調整したHAE発作頻度、期間で調整した1ヵ月あたりのレスキュー薬の使用頻度、C4タンパク量 等 |
解析計画
安全性の主要評価解析は、文書による同意/アセントの取得及びランダム化後に本剤を1回以上(又は一部)投与したすべての被験者で構成されるSafety Populationを用いて実施した。各評価項目で規定した安全性イベントのPTIRは、以下の通り算出した。
- 被験者に基づくPTIR=(各治療期に各安全性イベントが発現した被験者の総数)/(各被験者に最初の安全性イベントが発現した日付の合計−当該被験者の投与開始日+1日)/(365.25日)。各安全性イベントが発現しなかった被験者は分子に含めなかったが、被験者の試験参加時間の合計はリスク時間に含めた。
- イベントに基づくPTIR=(各治療期に記録された各安全性イベントの総数)/(各被験者の各イベントが回復した日付の合計−当該被験者の投与開始日+1日)/(365.25日)。各安全性イベントの発現の有無にかかわらず、被験者の試験参加時間の合計をリスク時間に含めた。
有害事象、注射部位反応及び注射部位反応以外の有害事象は、重症度、治験薬との因果関係、転帰及び重篤度を用量ごとに要約した。
有効性の評価項目は、Intent-to-Treat(ITT)Populationを用いて要約した。ITT Populationは、治験薬投与の有無に関わらず文書による同意/アセントを取得し、ランダム化されたすべての被験者で構成された。HAE発作を含むすべての有効性の評価項目は、併合したHAE発作を用いて主要評価項目の解析として解析した。期間で調整したHAE発作頻度は、HAE発作回数を本剤の期間で除して被験者ごとに算出し、用量ごとに要約統計量で要約した。レスポンダー及びノンレスポンダーの被験者数と割合、並びにレスポンダーの割合での40 IU/kg投与と60 IU/kg投与との差を要約し、40 IU/kg投与、60 IU/kg投与及び本剤併合集団でのレスポンダーの割合に対しWilsonの95%信頼区間を算出した。
6. 用法及び用量
本剤を添付の溶解液全量で溶解し、皮下投与する。通常、1回体重1kg当たり60国際単位を週2回投与する。
患者背景(Safety Population)
| 40 IU/kg投与群 (N=63) | 60 IU/kg投与群 (N=70) | 全体 (N=126) | ||
|---|---|---|---|---|
| 年齢、歳 | 平均値[標準偏差] | 40.8[14.96] | 40.8[16.01] | 40.5[15.56] |
| 最小値,最大値 | 8,67 | 10,72 | 8,72 | |
| 中央値 | 43.0 | 41.5 | 41.0 | |
| 性別、n(%) | 女性 | 40(63.5%) | 41(58.6%) | 76(60.3%) |
| 男性 | 23(36.5%) | 29(41.4%) | 50(39.7%) | |
| 人種、n(%) | 白人 | 60(95.2%) | 67(95.7%) | 121(96.0%) |
| 黒人又はアフリカ系米国人 | 1(1.6%) | 1(1.4%) | 2(1.6%) | |
| アジア人 | 0 | 1(1.4%) | 1(0.8%) | |
| その他 | 2(3.2%) | 1(1.4%) | 2(1.6%) | |
| 体重(kg) | 平均値[標準偏差] | 86.05[23.270] | 84.91[24.603] | 85.16[23.679] |
| 最小値,最大値 | 45.5,143.2 | 29.4,148.6 | 29.4,148.6 | |
| 中央値 | 87.20 | 80.00 | 83.00 | |
| BMI(kg/m2) | 平均値[標準偏差] | 29.62[6.919] | 29.00[7.428] | 29.21[7.230] |
| 最小値,最大値 | 16.7,47.1 | 15.1,54.2 | 15.1,54.2 | |
| 中央値 | 29.36 | 27.93 | 28.29 | |
BMI:体格指数
6. 用法及び用量
本剤を添付の溶解液全量で溶解し、皮下投与する。通常、1回体重1kg当たり60国際単位を週2回投与する。
安全性
人年法による発現率(PTIR) 主要評価項目
被験者に基づくPTIRは、全体で抗C1-INH抗体0.06例/人年、試験中止に至った有害事象0.02例/人年、血栓塞栓性事象及び入院を要したHAE発作各0.01例/人年であり、イベントに基づくPTIRは、全体で抗C1-INH抗体0.07件/人年、試験中止に至った有害事象0.02件/人年、血栓塞栓性事象及び入院を要したHAE発作各0.01件/人年でした。
重度の注射部位反応、本剤との因果関係ありと判定された重篤な有害事象及びアナフィラキシー関連事象は認められませんでした。
被験者及びイベントに基づく人年法による発現率(PTIR)(Safety Population)
| 40 IU/kg投与群 (N=63) | 60 IU/kg投与群 (N=70) | 全体 (N=126) | ||||
| PTIR | (95%CI) | PTIR | (95%CI) | PTIR | (95%CI) | |
| 被験者に基づくPTIR(例/人年) | ||||||
| 試験の早期中止に至った有害事象 | 0.01 | (<0.005,0.07) | 0.03 | (0.01,0.09) | 0.02 | (<0.01,0.06) |
| 血栓塞栓性事象 | 0.00 | − | 0.01 | (<0.005,0.06) | 0.01 | (<0.005,0.03) |
| アナフィラキシー | 0.00 | − | 0.00 | − | 0.00 | − |
| 入院を要したHAE発作 | 0.00 | − | 0.00 | − | 0.01 | (<0.005,0.03) |
| 重度の注射部位反応 | 0.00 | − | 0.00 | − | 0.00 | − |
| 本剤との因果関係ありと判定された 重篤な有害事象 | 0.00 | − | 0.00 | − | 0.00 | − |
| 抗C1-INH抗体(阻害抗体又は非阻害抗体)※ | 0.06 | (0.02,0.14) | 0.06 | (0.02,0.14) | 0.06 | (0.03,0.10) |
| イベントに基づくPTIR(件/人年) | ||||||
| 試験の早期中止に至った有害事象 | 0.01 | (<0.005,0.07) | 0.03 | (0.01,0.09) | 0.02 | (<0.01,0.05) |
| 血栓塞栓性事象 | 0.00 | − | 0.01 | (<0.005,0.06) | 0.01 | (<0.005,0.03) |
| アナフィラキシー | 0.00 | − | 0.00 | − | 0.00 | − |
| 入院を要したHAE発作 | 0.00 | − | 0.00 | − | 0.01 | (<0.005,0.03) |
| 重度の注射部位反応 | 0.00 | − | 0.00 | − | 0.00 | − |
| 本剤との因果関係ありと判定された 重篤な有害事象 | 0.00 | − | 0.00 | − | 0.00 | − |
| 抗C1-INH抗体(阻害抗体又は非阻害抗体)※ | 0.06 | (0.02,0.14) | 0.09 | (0.04,0.17) | 0.09 | (0.04,0.13) |
40 IU/kgから60 IU/kgへ増量した7例はSafety Populationの両方の投与群に含めた。
11.1 重大な副作用
11.1.1 ショック、アナフィラキシー(いずれも頻度不明)
貧血、血圧上昇、血圧低下、潮紅、じん麻疹、呼吸困難、頭痛、めまい、悪心等がみとめられた場合には
直ちに投与を中止し、適切な処置を行うこと。なお、アナフィラキシーは遺伝性血管性浮腫の発作と
同様の症状を示すため、観察を十分に行うこと。
6. 用法及び用量
本剤を添付の溶解液全量で溶解し、皮下投与する。通常、1回体重1kg当たり60国際単位を週2回投与する。
有害事象 副次評価項目
有害事象は40 IU/kg投与群で63例中56例(88.9%)、60 IU/kg投与群で70例中58例(82.9%)に発現し、副作用は40 IU/kg投与群で63例中36例(57.1%)、60 IU/kg投与群で70例中32例(45.7%)に発現しました。
主な副作用は、40 IU/kg投与群で注射部位疼痛が63例中17例(27.0%)、注射部位紅斑が63例中10例(15.9%)、注射部位内出血が63例中9例(14.3%)に発現し、60 IU/kg投与群で注射部位紅斑が70例中12例(17.1%)、注射部位疼痛 が70例中10例(14.3%)、注射部位反応が70例中8例(11.4%)に発現しました。
重篤な有害事象は、40 IU/kg投与群で4例(びまん性大細胞型B細胞性リンパ腫1例、挫傷1例、脱水及び低カリウム血症1例、気管支炎1例)、60 IU/kg投与群で5例(胆石症1例、複視1例、急性心筋梗塞1例、浮動性めまい及び胸痛1例、肺炎1例)に認められました。試験中止に至った有害事象は、40 IU/kg投与群で1例(筋肉痛)、60 IU/kg投与群で3例(頭痛1例、急性心筋梗塞1例、関節痛1例)に認められました。死亡例は認められませんでした。
有害事象の概要(Safety Population)
| 40 IU/kg投与群 (N=63) | 60 IU/kg投与群 (N=70) | 全体 (N=126) | |
|---|---|---|---|
| 有害事象 | 56(88.9%) | 58(82.9%) | 108(85.7%) |
| 副作用 | 36(57.1%) | 32(45.7%) | 66(52.4%) |
| 重篤な有害事象 | 4(6.3%) | 5(7.1%) | 9(7.1%) |
| 試験中止に至った有害事象 | 1(1.6%) | 3(4.3%) | 4(3.2%) |
| 死亡 | 0 | 0 | 0 |
40 IU/kgから60 IU/kgへ増量した7例はSafety Populationの両方の投与群に含めた。
例数(%)
6. 用法及び用量
本剤を添付の溶解液全量で溶解し、皮下投与する。通常、1回体重1kg当たり60国際単位を週2回投与する。
有効性
期間で調整したHAE発作頻度 探索的評価項目
期間で調整したHAE発作頻度の平均値(標準偏差)は、1ヵ月あたりでは60 IU/kg投与群において0.45(0.858)回/月であり、HAE発作頻度の中央値(第1四分位、第3四分位)は、60 IU/kg投与群において0.09(0.00、0.52)回/月でした。
1年あたりの期間で調整したHAE発作頻度の平均値(標準偏差)は、60 IU/kg投与群において5.45(10.299)回/年であり、HAE発作頻度の中央値(第1四分位、第3四分位)は、60 IU/kg投与群において1.02(0.00、6.21)回/年でした。
期間で調整したHAE発作頻度(ITT Population)
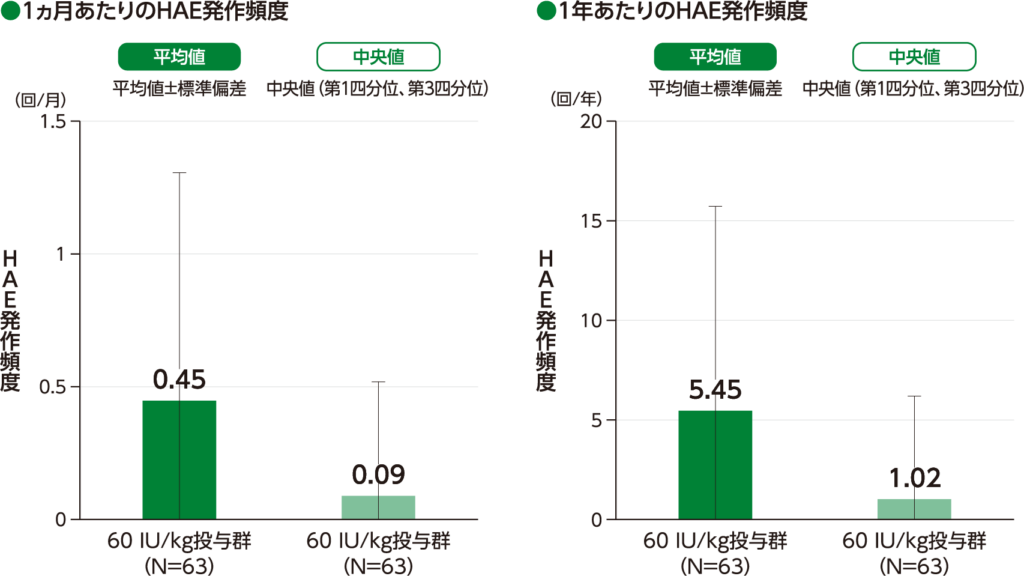
・40 IU/kg投与群の結果は、承認外の用法及び用量のため削除した。
6. 用法及び用量
本剤を添付の溶解液全量で溶解し、皮下投与する。通常、1回体重1kg当たり60国際単位を週2回投与する。
レスポンダーの割合 副次評価項目
期間で調整したHAE発作頻度が、試験参加時の適格性基準として用いたHAE発作頻度と比較して50%以上相対的に減少したレスポンダーの割合(95% Wilson信頼区間)は、60 IU/kg投与群で91.7(81.9、96.4)%(55/60例)でした。
期間で調整したHAE発作頻度が50%以上相対的に減少したレスポンダーの割合(ITT Population)
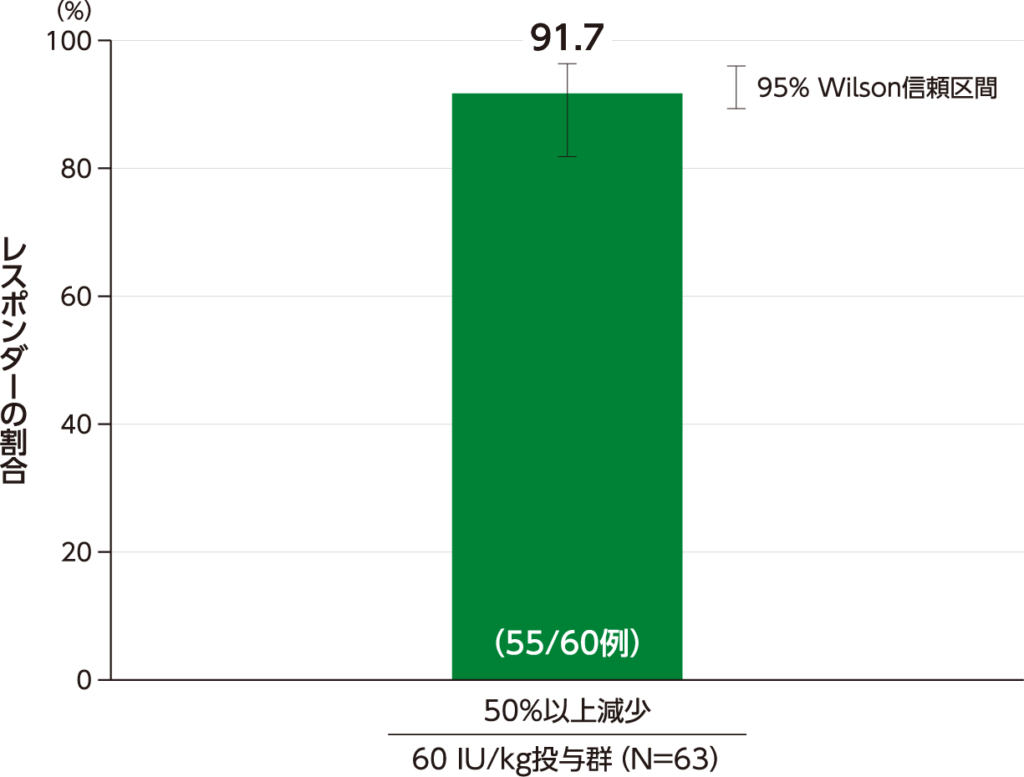
・40 IU/kg投与群の結果は、承認外の用法及び用量のため削除した。
6. 用法及び用量
本剤を添付の溶解液全量で溶解し、皮下投与する。通常、1回体重1kg当たり60国際単位を週2回投与する。
4週あたりの期間で調整したHAE発作頻度が1回未満へ減少した被験者の割合 副次評価項目
4週あたりの期間で調整したHAE発作頻度が1回未満へ減少した被験者の割合は、60 IU/kg投与群で85.7%(54/63例)でした
4週あたりの期間で調整したHAE発作頻度が1回未満へ減少した被験者の割合(ITT Population)
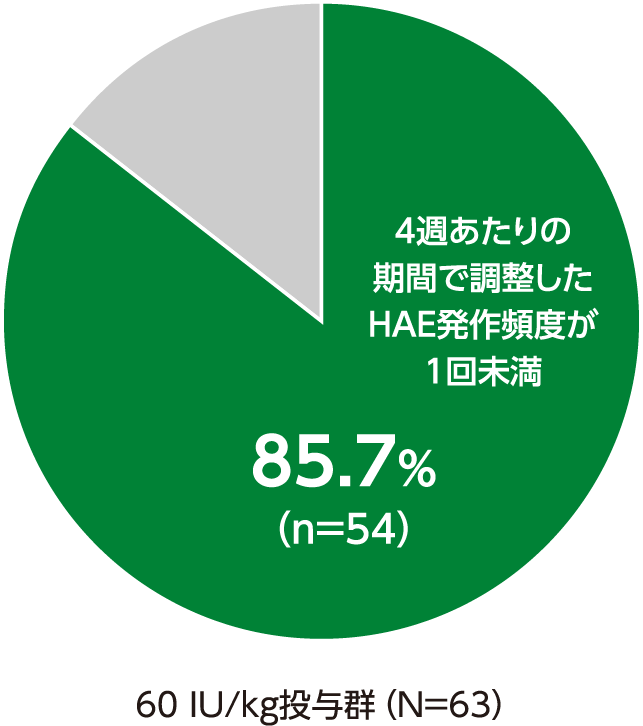
・40 IU/kg投与群の結果は、承認外の用法及び用量のため削除した。
6. 用法及び用量
本剤を添付の溶解液全量で溶解し、皮下投与する。通常、1回体重1kg当たり60国際単位を週2回投与する。
期間で調整したレスキュー薬の使用頻度 探索的評価項目
期間で調整したレスキュー薬の使用頻度の平均値(標準偏差)は、1ヵ月あたりでは60 IU/kg投与群において0.31(0.804)回/月であり、レスキュー薬の使用頻度の中央値(第1四分位、第3四分位)は、60 IU/kg投与群において0.00(0.00、0.25)回/月でした。
1年あたりの期間で調整したレスキュー薬の使用頻度の平均値(標準偏差)は、60 IU/kg投与群において3.76(9.648)回/年であり、レスキュー薬の使用頻度の中央値(範囲)は、60 IU/kg投与群において0.00(0.0、54.2)回/年でした。
期間で調整したレスキュー薬の使用頻度(ITT Population)
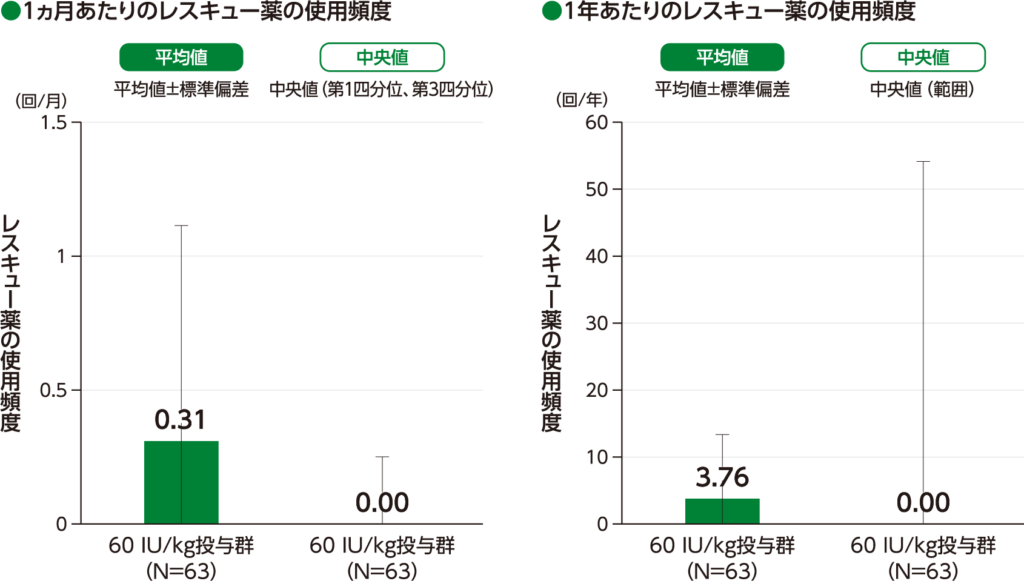
・40 IU/kg投与群の結果は、承認外の用法及び用量のため削除した。
6. 用法及び用量
本剤を添付の溶解液全量で溶解し、皮下投与する。通常、1回体重1kg当たり60国際単位を週2回投与する。
薬物動態/薬力学
C4タンパク量 探索的評価項目
ベースライン時のC4タンパク量の平均値(標準偏差)は60 IU/kg投与群で9.1(5.0)mg/dL、試験終了時のC4タンパク量の平均値(標準偏差)は60 IU/kg投与群で16.3(6.6)mg/dLでした。
C4タンパク量の平均値(±標準偏差)の推移
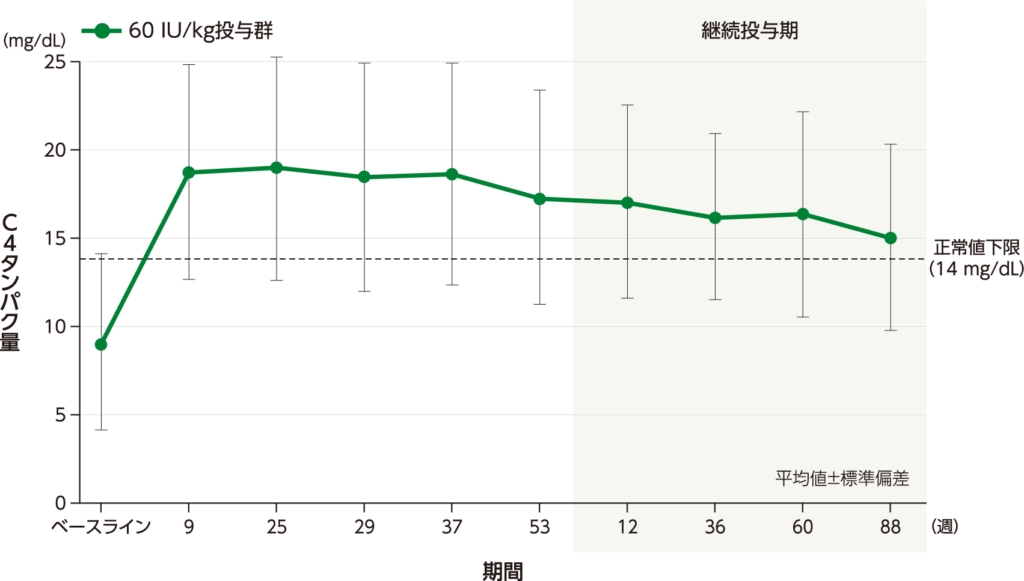
・40 IU/kg投与群の結果は、承認外の用法及び用量のため削除した。
6. 用法及び用量
本剤を添付の溶解液全量で溶解し、皮下投与する。通常、1回体重1kg当たり60国際単位を週2回投与する。